摘要: 肾细胞癌是常见且致命的疾病,占肾癌的绝大多数。肾细胞癌是一种异质性的复杂疾病,主要由三种组织学亚型组成,存在不同的生物学和临床差异。如今,科技的发展能够得到肾细胞癌的分子亚型和生物标志物。在这项研究中,我们将三种不同的特征选择技术进行组合,即mRMR、Lasso、Boruta,利用投票法的思路从TCGA多个单组学数据集中选择最显著的特征,并将其作为基础机器学习模型的输入,用于肾细胞癌组织学亚型分类。我们评估了六种不同的分类模型,包括逻辑回归(LR)、随机森林(RF)、支持向量机(SVM)、朴素贝叶斯(NB)、k-最近邻(KNN)和XGBoost。结果表明,基于应用本文的新特征选择流程,miRNA成熟链表达RNAseq数据集提供的特征在准确性方面优于其他分类方法,在逻辑回归模型下能达到0.9779的准确率与0.9834的AUC,取得了最高性能。因此,我们改进和细化的特征选择和分类提供了诊断标志物,可能有助于提高诊断的准确性,从而帮助设计早期治疗策略,提高肾细胞癌患者的生存率。
Abstract:
Renal cell carcinoma is a common and fatal disease, accounting for the majority of kidney cancers. There are three basic histological subtypes of renal cell carcinoma, each of which has unique biolog-ical and clinical characteristics, which represents a complex and heterogeneous ailment. The avail-ability of molecular subtypes and biomarkers for renal cell carcinoma is made possible by modern technological advancements. In this study, we combined three different feature selection tech-niques, namely mRMR, Lasso, and Boruta, using the idea of voting method to select the most signif-icant features from multiple single-omics datasets of TCGA and use them as input to a base machine learning model for histological subtype classification of renal cell carcinoma. We evaluated six clas-sification models, including logistic regression (LR), random forest (RF), support vector machine (SVM), naive Bayes (NB), k-nearest neighbor (KNN), and XGBoost. The results demonstrate that the features from the miRNA mature strand expression RNAseq dataset outperformed other classifica-tion methods based on the application of the new feature selection process in this paper, achieving the highest performance with an accuracy of 0.9779 and an AUC of 0.9834 under the logistic regres-sion model. As a result, the enhanced and refined feature selection and categorization offer diag-nostic indicators that could help increase the accuracy of diagnosis, aid in the development of early treatment plans, and enhance the survival of patients with renal cell carcinoma.
1. 引言
肾细胞癌(RCC)是肾小管上皮细胞的异质性癌症,占肾脏癌症的90%以上,一种常见且致命的疾病 [1] [2] 。根据世界卫生组织的最新数据,每年肾癌新增约430,000例,死亡近180,000例 [3] 。肾细胞癌由许多不同的癌症亚型组成,每个亚型都有不同的组织学、不同的遗传和分子改变、不同的临床病程以及不同的治疗反应 [4] 。RCC组织学亚型主要包括以下三类:肾透明细胞癌(ccRCC)、乳头状肾细胞癌(pRCC)和嫌色细胞癌(chRCC),其中肾透明细胞癌是最为常见的亚型,约占肾细胞癌的75%,乳头状肾细胞癌占15%~20%,嫌色肾细胞癌约占5% [2] 。组织病理学评估是诊断ccRCC、pRCC和chRCC的金标准。精确判别并了解各个亚型之间的差异对于改进患者的管理和治疗至关重要。
随着基因组学和转录组学技术的快速发展,越来越多的基因组数据被收集起来供人研究。癌症基因组图谱(TCGA, https://tcga-data.nci.nih.gov/tcga/)是一个里程碑式的癌症基因组学项目,它旨在描述驱动不同癌症的遗传特征 [5] 。TCGA包含不同类型的组学数据,包括转录组数据(mRNA、lncRNA和miRNA)、基因组数据(突变、CNV)、表观基因组数据(DNA甲基化)、蛋白质组数据和临床信息数据 [5] [6] 。许多研究仅采用全向组学数据进行癌症分类研究,最常见的是基因表达数据 [7] 。TCGA中包含三个关于肾脏癌症的项目,并分配了四个字母的代码以反映其组织学定义:KICH (肾嫌色细胞癌)、KIRC (肾透明细胞癌)和KIRP (肾乳头状细胞癌)。
在这项研究中,我们旨在充分利用TCGA数据,获得能够准确区分肾细胞癌进行亚型的特征,以确定共有的和亚型特异性分子特征,这将为开发RCC的疾病特异性治疗方法和预后生物标志物提供基础。我们改进的分类可以带来更准确的诊断和更适合患者的护理,本文流程也可以应用于其他癌症或疾病。
2. 材料与方法
2.1. 患者数据
肾细胞癌组学数据来自TCGA中的肾嫌色细胞癌、肾透明细胞癌以及肾乳头状细胞癌三个队列的组合(KICH,KIRC,KIRP,https://tcga.xenahubs.net)。为了对肾细胞癌进行分类,我们考虑了包括基因级拷贝数变异、外显子表达RNA测序、甲基化数据、基因表达RNA测序和miRNA成熟链表达RNA测序在内的五种单组学数据集的数据来进行探究。我们在下文中用copynumber、exon RNA-seq、methylation、gene RNA-seq和miRNA的缩写来指这五个数据集。生存分析数据由表型数据集提供。

Table 1. Original samples and feature counts for each data set of renal cell carcinoma
表1. 肾细胞癌各个数据集原始样本与特征数
五种单组学数据各自进行亚型赋值,新创建“subtype”列并将“KICH”、“KIRC”、“KIRP”三种亚型分别赋予0、1、2。然后我们再对其进行数据合并,得到表1所述的五个合并后的数据集。接下来分别对其进行去除缺失值、均值插补和标准化,以确定合并数据是否有利于肾细胞癌的组织学亚型分类。其中,每个数据集的20%的数据被留作独立的测试集,80%的数据被用于训练。
2.2. 数据预处理与特征选择
当数据规模有限但维度较高时,过拟合等问题容易导致分类精度降低。因此,选择合适的特征选择方法将使我们更有效地分析数据。在本文中,首先删除缺失超过30%的列。接下来每个数据集的低方差的特征(methylation450k的方差 < 0.05,copynumber、miRNA与gene RNA-seq的方差 < 0.5,exon RNA-seq的方差 < 1)被剔除,因为它们对分类的作用不大。我们同时考虑了各个特征与组织学亚型的相关性,研究发现,去除亚型相关性低的变量(所有数据集的spearman相关系数的绝对值 < 0.1)对本文的研究产生了些许的提升。
接下来对数据集进行数据标准化,为了特征选择的鲁棒性,我们同时选择LASSO [8] 、mRMR (Max-Relevance and Min-Redundancy) [9] 和Boruta [10] 进行特征选择。当某一特征在一次交叉验证中被三种特征选择方法同时选中两次或两次以上时,则认为该特征对肾细胞癌的亚型分类有意义。5折交叉验证经过上述程序后都能够选出的特征即是最终的特征,并将其输入到多个机器学习模型中,判断哪个数据集最能将肾细胞癌的亚型精确分类。虽然原始特征的数量和每个数据集所包含的信息量差别很大,但为了更加明确地看出哪个数据集最能对肾细胞癌亚型分类提供信息,我们特意固定了交叉验证中每次mRMR选择的特征数量(所有数据集均选择50个特征)。每个数据集的最终特征显示在表3中。
2.3. 机器学习模型
如上所述,我们考虑将交叉验证重叠的特征作为机器学习模型的输入。在本文中,我们将平均准确率和平均AUC作为判断亚型预测好坏的标准。其中,准确率是通过预测结果与表型数据集中的表达亚型的一致性来评估的,AUC定义为ROC曲线下坐标轴所包围的区域。观察不同交叉验证下的准确率和AUC的标准差,有助于判断该输入特征下的机器学习模型是否稳健。
在这项研究中,我们选择六种机器学习模型:逻辑回归(LR)、随机森林(RF)、支持向量机(SVM)、朴素贝叶斯(NB)、k近邻(KNN)和XGBoost。这些基础的机器学习模型被应用于根据选定的特征估计肾细胞癌的组织学亚型,以确定本文的特征选择方法是否有助于癌症亚型的分类。所有代码都可以通过python轻松实现。
3. 数据可用性
支持本研究结果的样本数据由TCGA中的KICH,KIRC,KIRP中公开提供(https://tcga.xenahubs.net)。
4. 结论
4.1. 特征选择
将KICH、KIRC、KIRP三种亚型合并,分别得到copynumber、exon RNA-seq、methylation、gene RNA-seq和miRNA五个总数据集,具体样本与特征数目如表2所示,此处只选择肾细胞癌亚型分类准确率与AUC最高的一组数据进行详细说明,即miRNA数据集。我们得到721个样本,共有1847个特征,是合并后的五个数据集中样本与特征数量最少的数据集。其中每个亚型的数量虽有一定差别,但仍较为平衡,不需要考虑过采样或欠采样:89个KICH,311个KIRC,321个KIRP。基于训练集与测试集的划分比例,在5折交叉验证中,每次交叉验证产生577个训练集和144个测试集。为了发现对准确划分亚型提供帮助的特征,近年来广泛采用三种特征选择技术:mRMR、LASSO和Boruta。
使用方法章节中讨论过的特征选择过程,我们最终从miRNA数据集中得到了77个特征,如表3所示。其余数据集所得特征与数量也于表3所示。各数据集选择出的特征的表达水平以及热力图由附录中的附图1与附图2展示,我们可以看出,使用本文特征选择流程挑选出的特征在三种亚型中差异明显,也可以反映出特征对肾细胞癌亚型分类有益处。
Table 3. Features extracted from each dataset
表3. 各数据集提取出的特征
4.2. 机器学习模型结果
为了评估本文的数据整合和特征选择过程是否有助于肾细胞癌的亚型分类,我们对合并后的各个数据集进行5折交叉验证,将上述特征选择流程得到的重要特征放入六个基础的机器学习模型中,并观察其最终的准确率与AUC。
肾细胞癌亚型分类的结果如图1所示。根据结果,我们可以清楚地看到,来自miRNA数据集的特征在所有数据中取得了最高的AUC,这与表2的结果一致,miRNA原始的特征最少,但共挑选出了最多的77个特征。来自gene RNA_seq数据集的特征对肾细胞癌亚型准确分类的贡献最小,但从图1中可以看出标准差很小,这表明gene RNA_seq可以为癌症亚型分类提供更稳定的特征。相比之下,其他三个数据集的标准差较大,在每个机器学习模型下的稳定性相对较差。
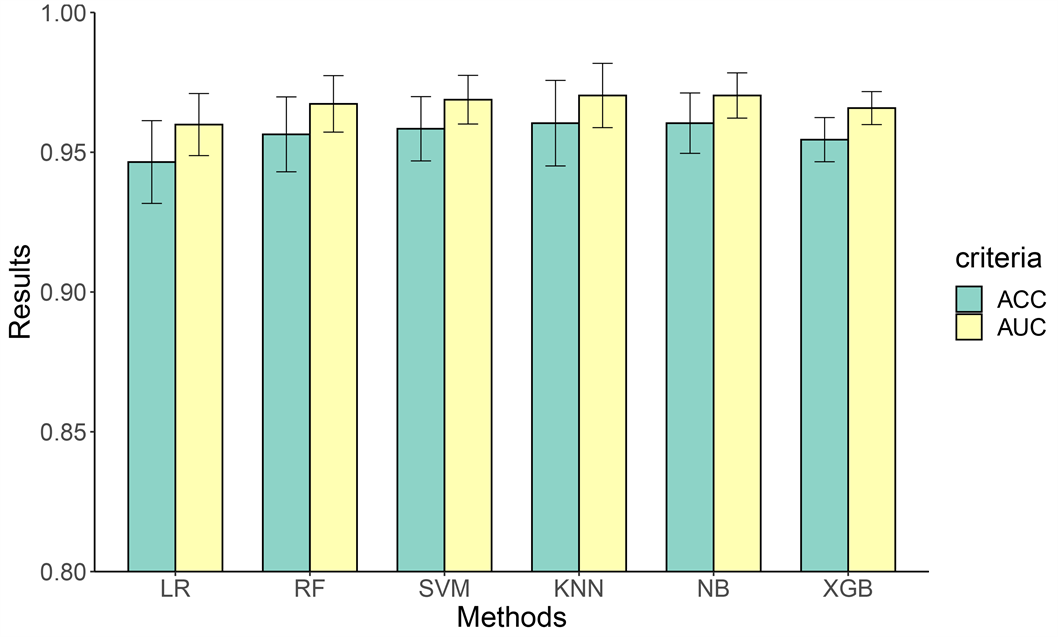
Figure 2. Mean accuracy and AUC of miRNA dataset
图2. miRNA数据集平均准确率和AUC
我们选择图1中的最优结果进行更深一步的调查,即miRNA数据集的结果。图2和表4充分表明,大多数机器学习方法在这种组合下产生的特征可以获得0.95甚至更高的AUC,最高甚至超过0.98。除了KNN和XGB这两种机器学习方法有偏大的标准差外,所有方法得到的准确率和AUC都比较稳定,特别是LR与SVM方法。在所有方法中,LR的结果最好,ACC为0.9779,AUC为0.9834。
Table 4. Results of using the new feature selection process with the machine learning classifier
表4. 使用新特征选择流程配合机器学习分类器的结果
5. 讨论
肾细胞癌是一种高度异质性的疾病,不同组织学亚型适合不同的治疗方案,且存在相差较大的预后。本文所述的研究对肾细胞癌至关重要。
本研究的创新之处在于,我们使用了多个TCGA单组学数据进行比较,应用了更全面的特征选择流程,采用多种类型的机器学习方法对肾细胞癌亚型进行分类,并评估每类的生存意义。在我们的研究中,最核心的内容是提出了一个同时使用mRMR、lasso和boruta的特征提取器,用于基于The Cancer Genome Atlas (TCGA)组学数据的肾细胞癌组织学亚型分类,利用六个基础的机器学习模型产生的准确率与AUC来判断最终亚型分类结果的优劣。虽然使用单一的特征选择方法提取特征很简单快捷,但结果很可能不尽人意。因此,我们决定采用更加鲁棒的投票法,结合三种特征选择技术,选择有利于肾细胞癌亚型分类的特征。最后,我们选择了miRNA数据集中的77个特征,使用六种机器学习模型评估特征选择技术的性能:逻辑回归(LR)、随机森林(RF)、支持向量机(SVM)、朴素贝叶斯(NB)、k近邻(KNN)和XGBoost,且每个机器学习模型都能获得出色的结果。其中,LR在肾细胞癌亚型分类中能够产生最高和最稳定的准确率和AUC。
图1中显示,miRNA数据集为肾细胞癌亚型分类提供了最强的鉴别力,即交叉验证下各个机器学习方法均获得最高的平均结果,methylation数据集的分类性能次之,gene RNA_seq数据集的结果虽不名列前茅,但最为稳定。
为了验证本文方法的稳健性,我们选择了肺腺癌和乳腺癌的TCGA数据进行验证。我们发现,虽然肾细胞癌多组学数据没有明显提高亚型分类的准确率与AUC,但使用多组学数据时肺腺癌的AUC高达0.97,乳腺癌的AUC也可以达到0.9以上,相较单组学数据皆存在显著提升,这也反映出本文提出的特征提取方法较为鲁棒,可以适用于各种癌症。
虽然我们的方法在肾细胞癌组织学亚型分类中取得了良好的效果,但也有一些局限性:1) 在我们的研究中,我们只研究了肾细胞癌,且选取了肺腺癌与乳腺癌进行验证,我们的方法是否可以扩展到研究其他癌症的亚型,还需要进一步探讨。2) 虽然合并这五个数据集没有对肾细胞癌组织学亚型分类产生更优的结果,但我们没有使用更全面的多组学数据进行亚型分类,目前还不知道是否有更好的TCGA数据组合。
附录
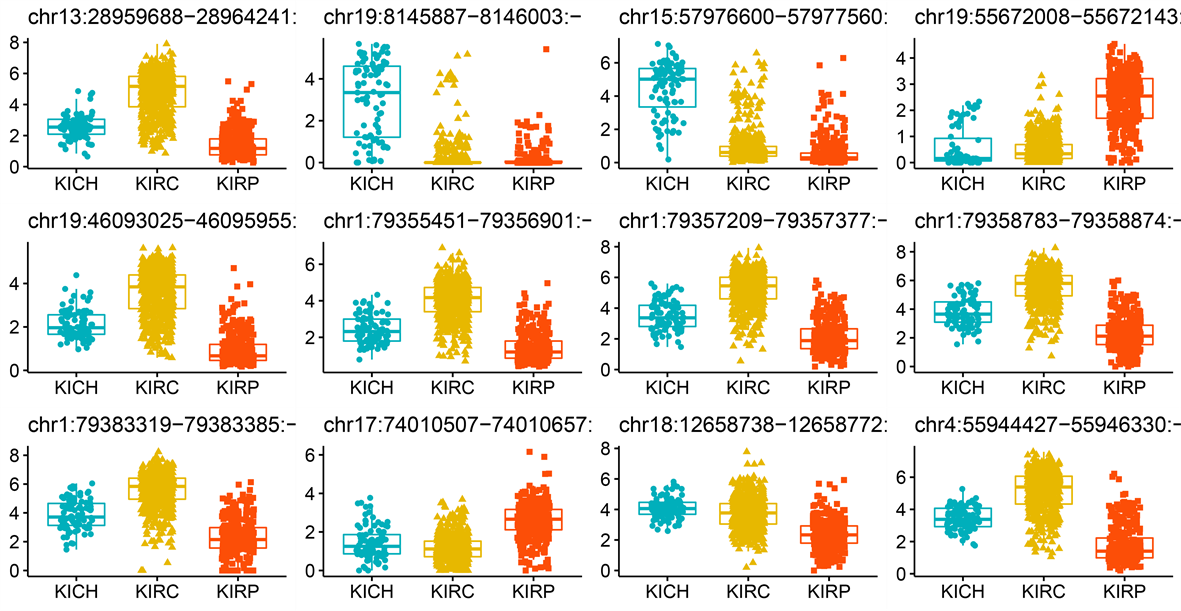

(a) 外显子表达水平
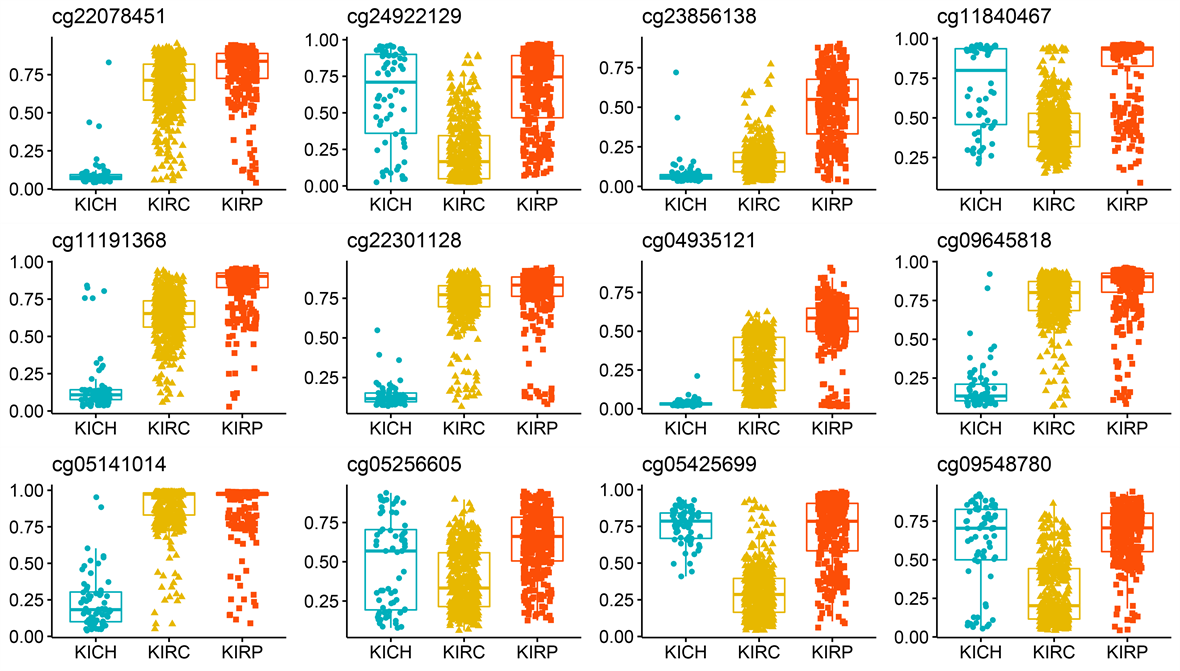
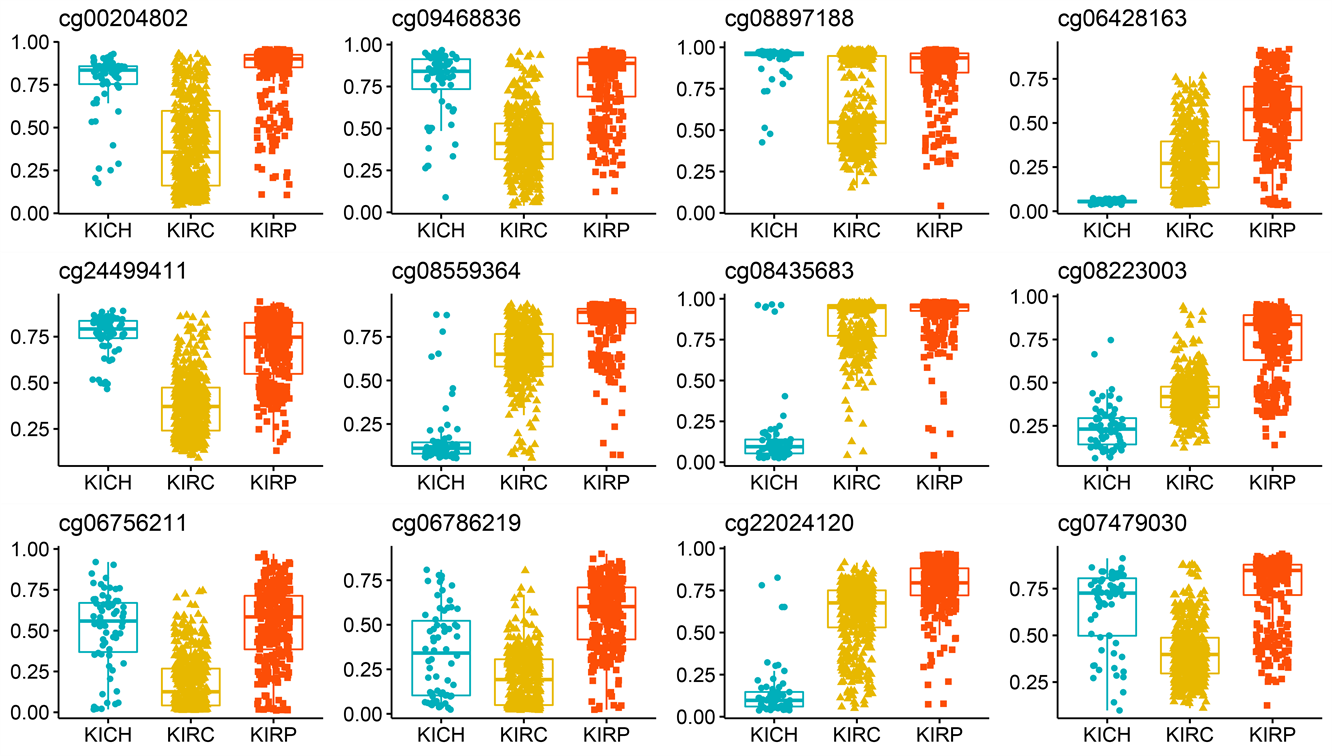
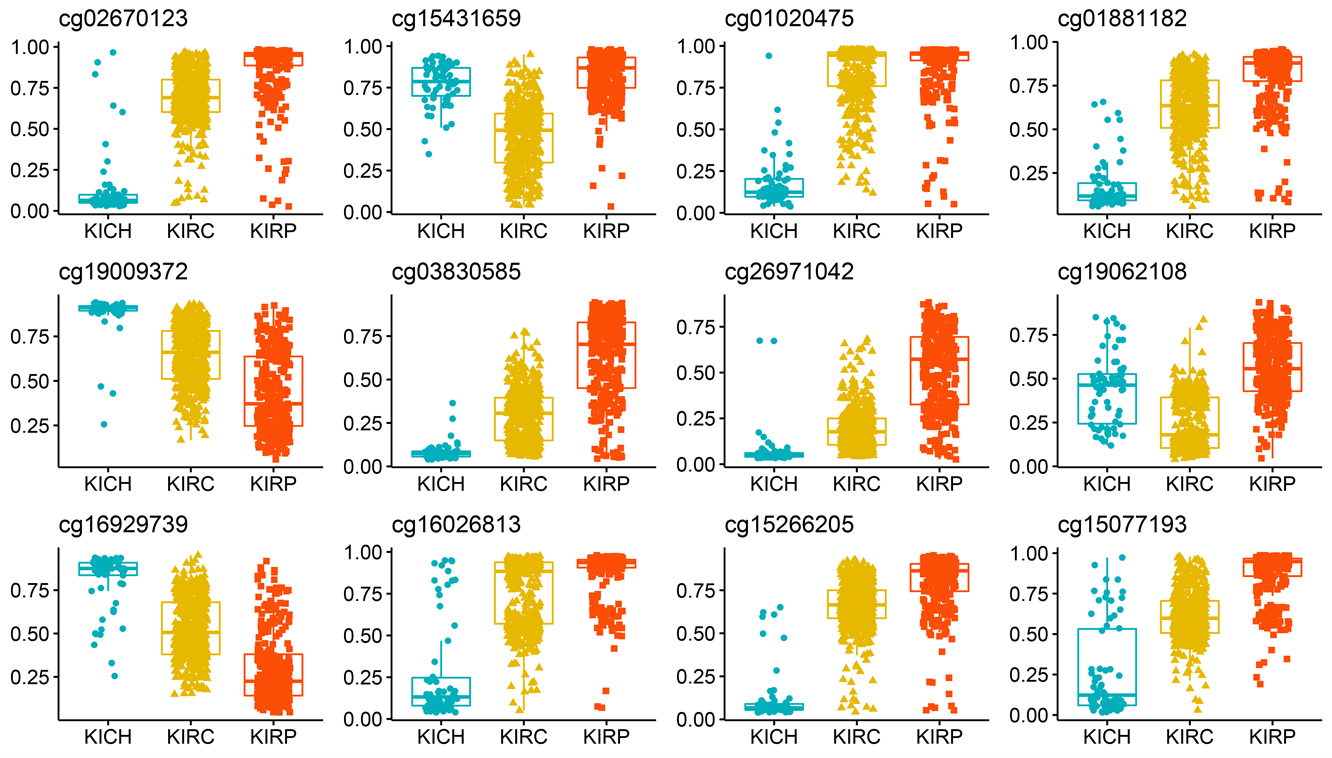

(b) 甲基化水平

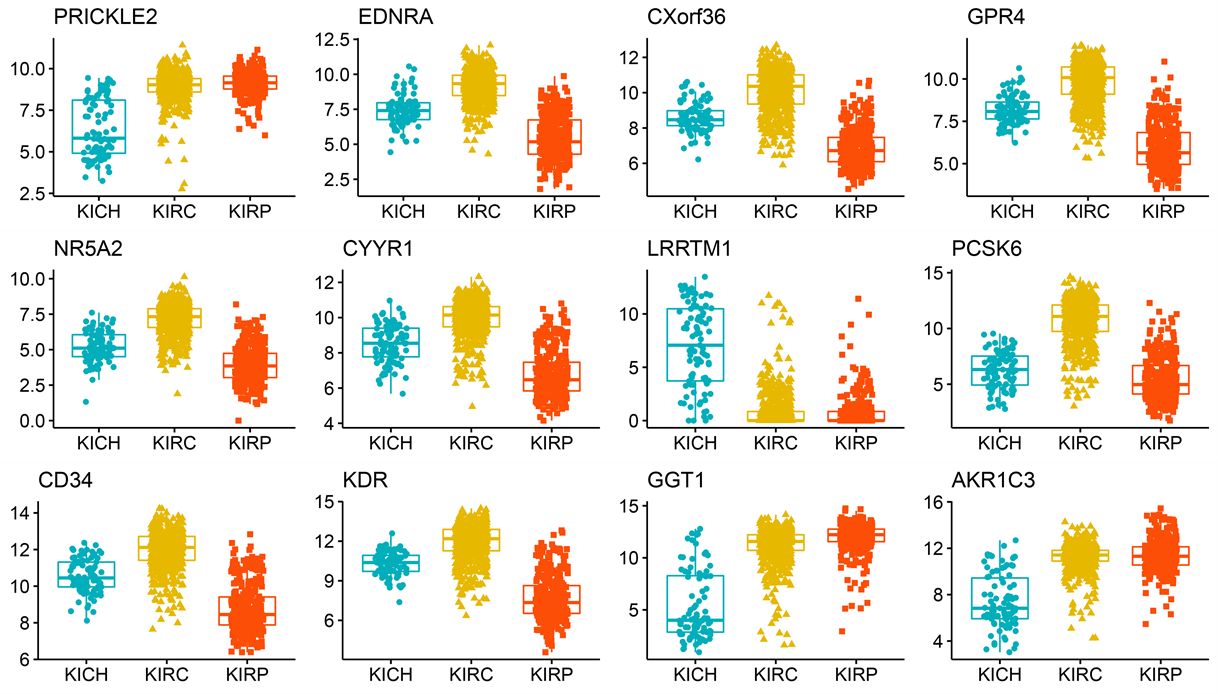
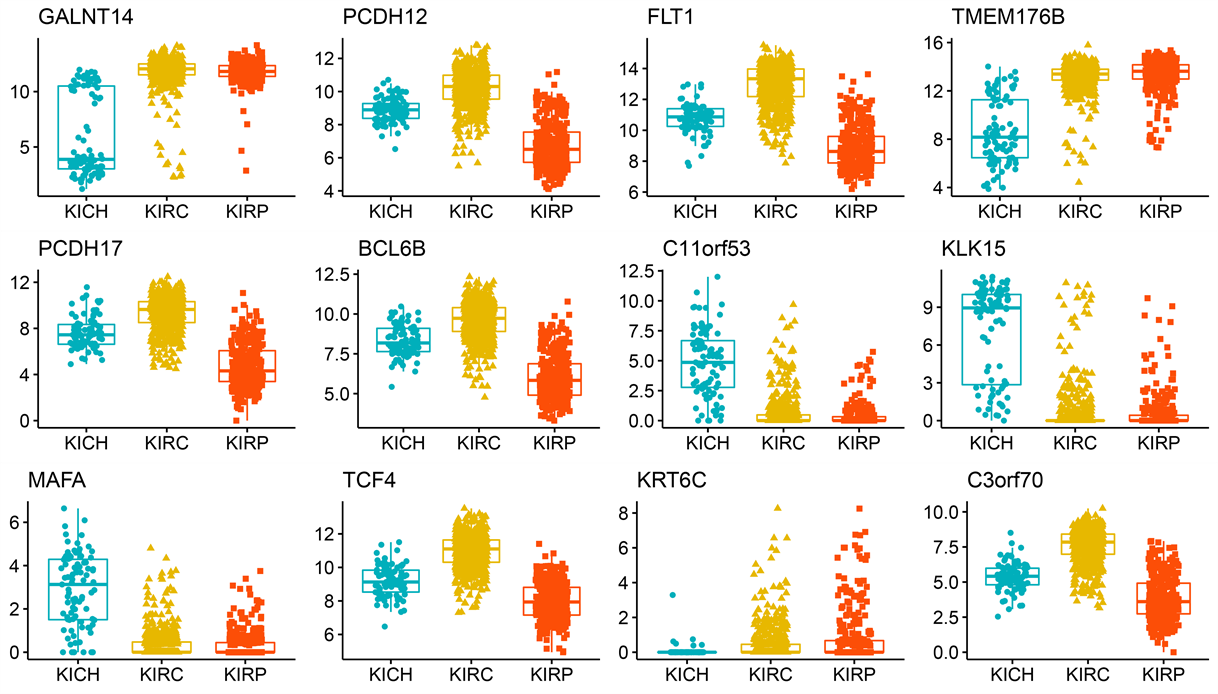
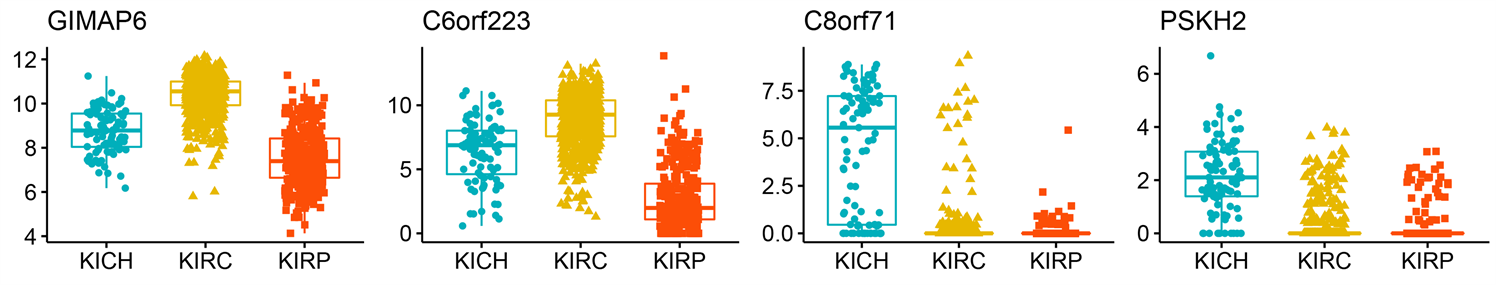
(c) RNA-seq表达水平
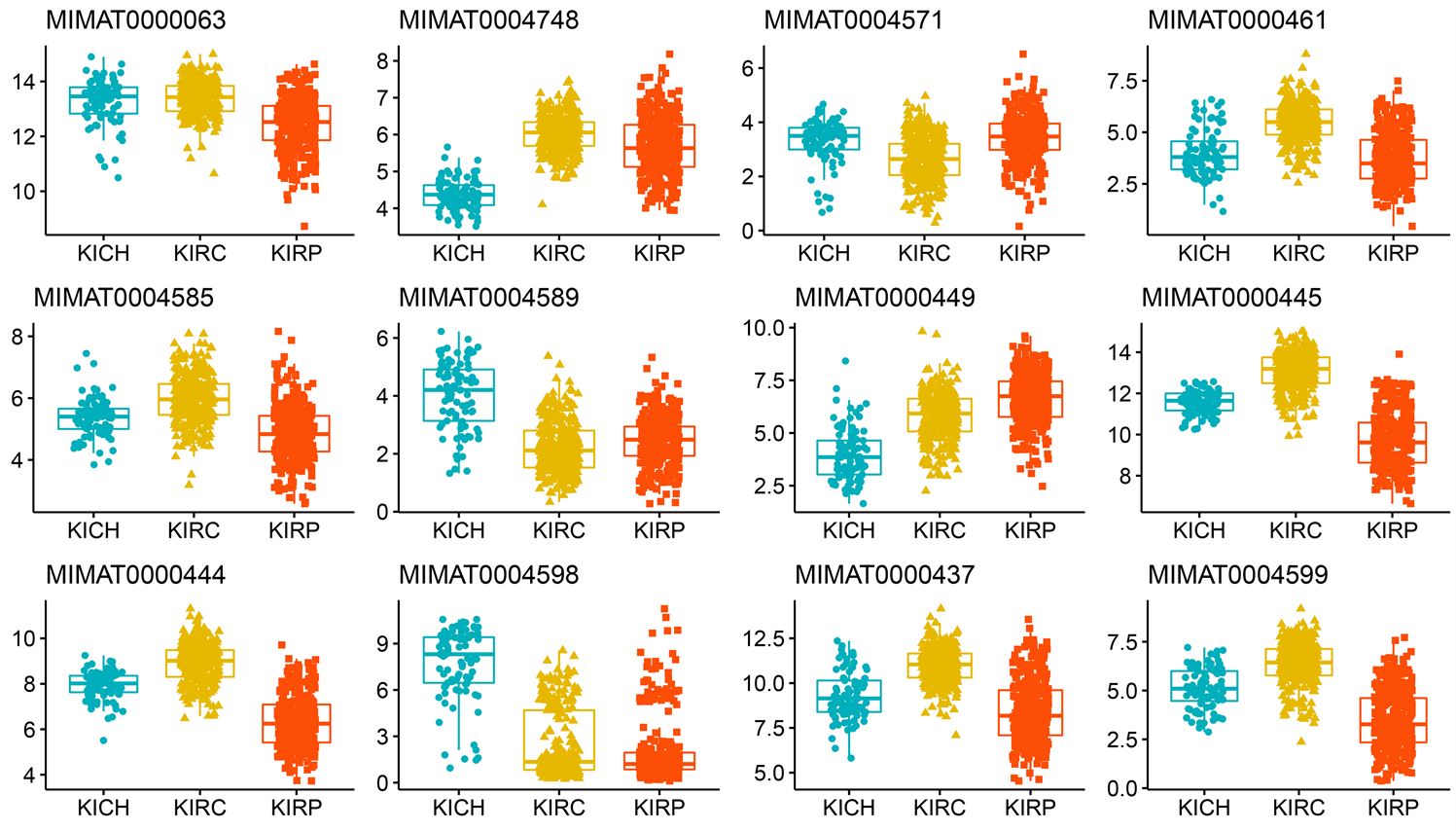
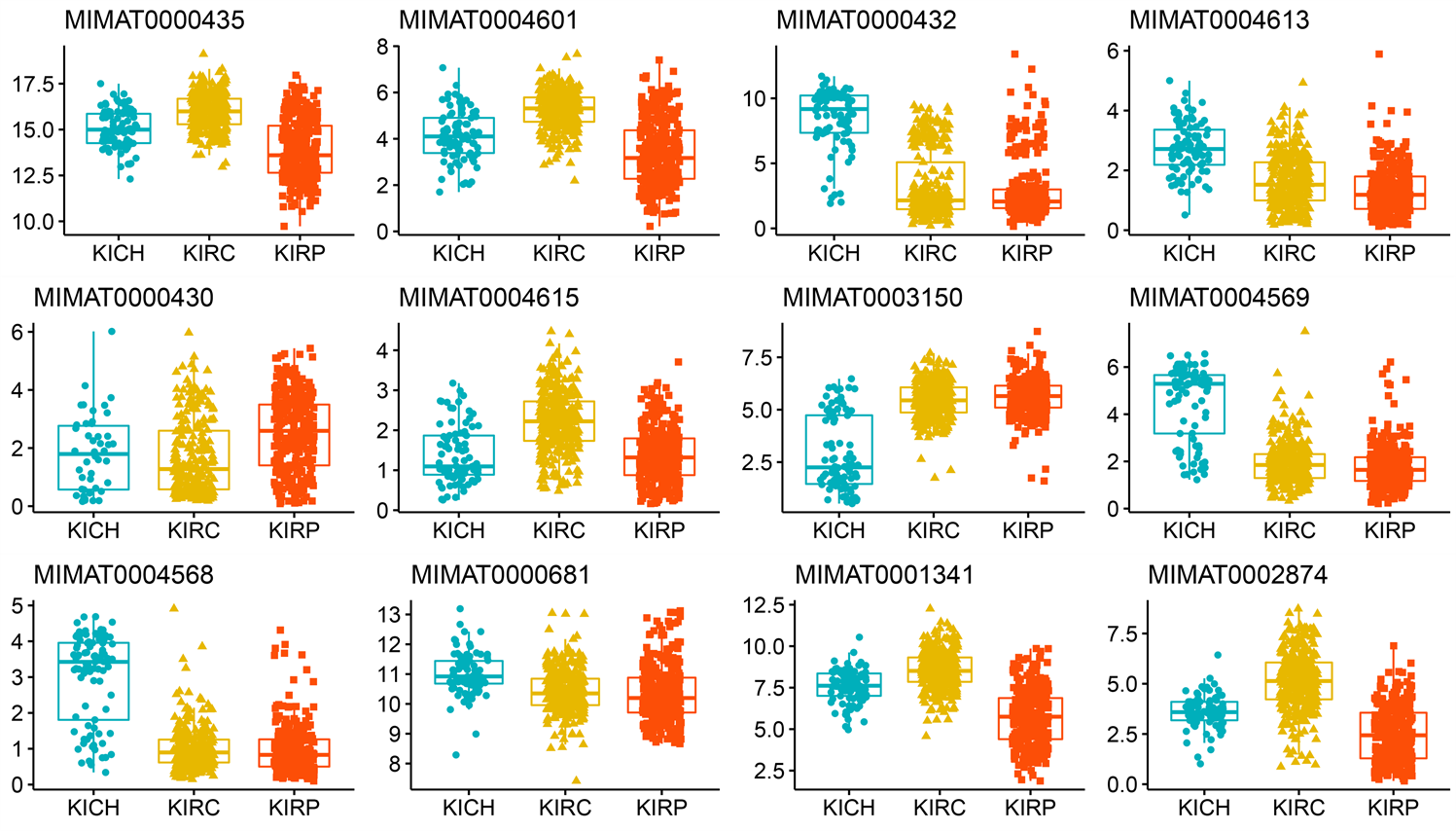
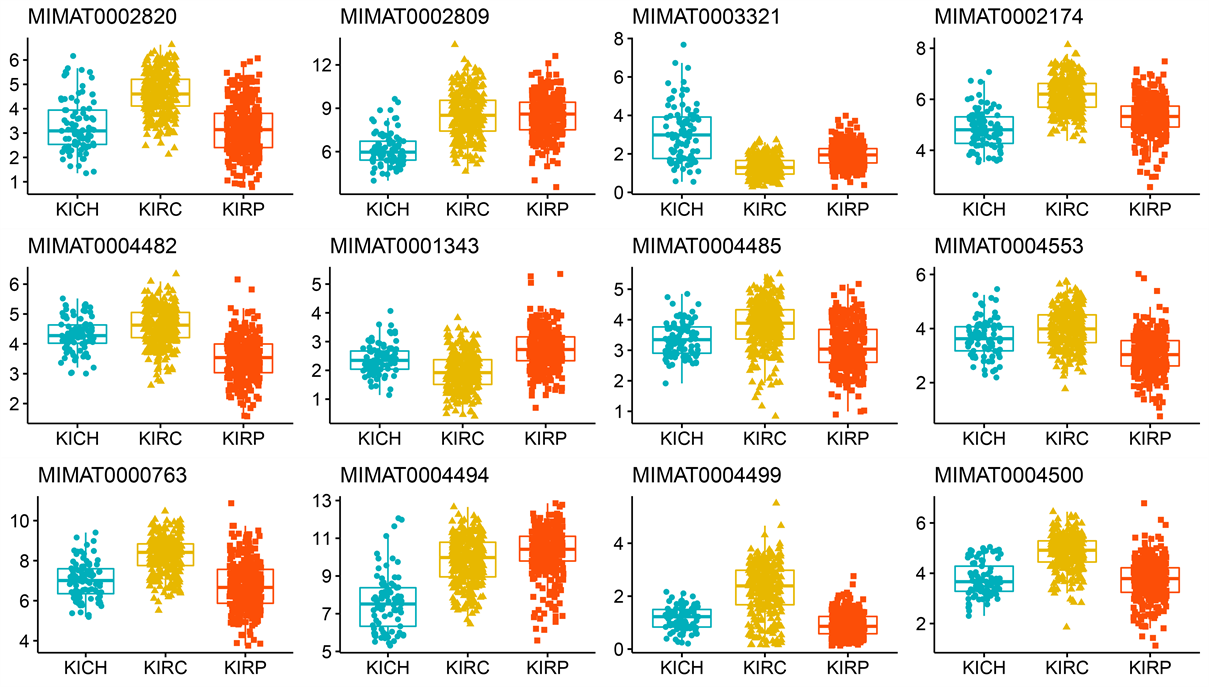

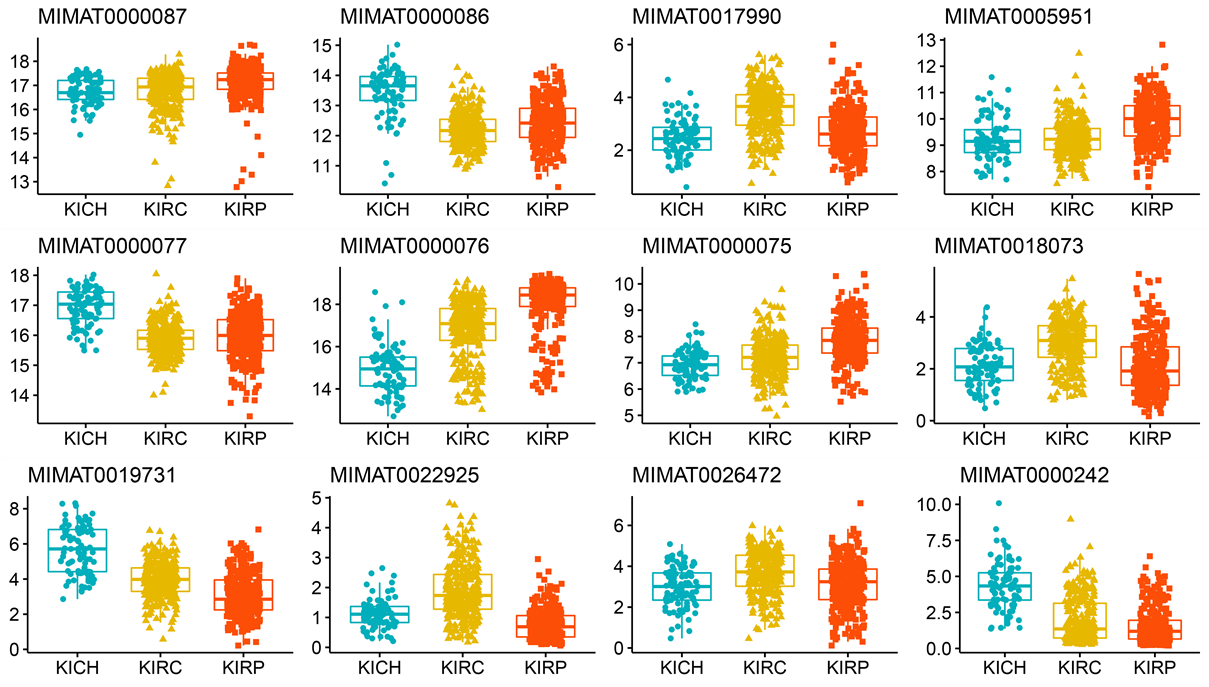
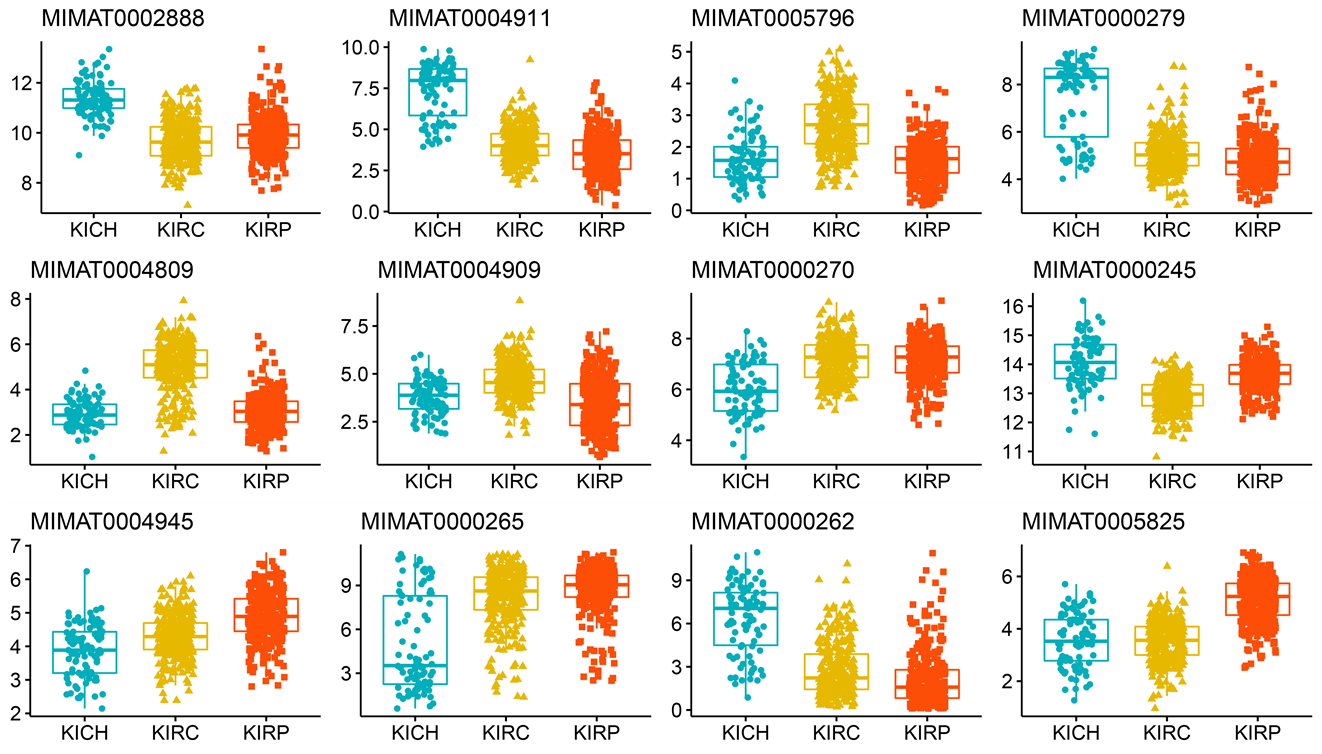
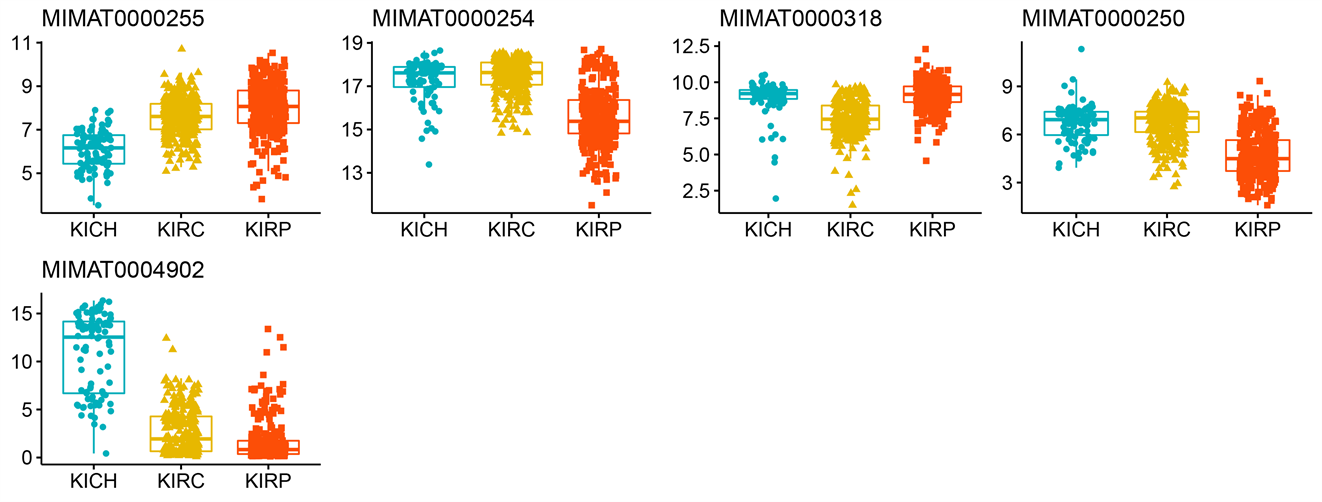 (d) miRNA成熟链表达水平
(d) miRNA成熟链表达水平
Figure S1. Box plots of expression levels of selected important features: exon expression level (a), methylation level (b), RNA-seq expression level (c), miRNA mature strand expression level (d)
附图1. 挑选出的重要特征的表达水平箱线图:外显子的表达水平(a),甲基化水平(b),RNA-seq的表达水平(c),miRNA成熟链表达水平(d)
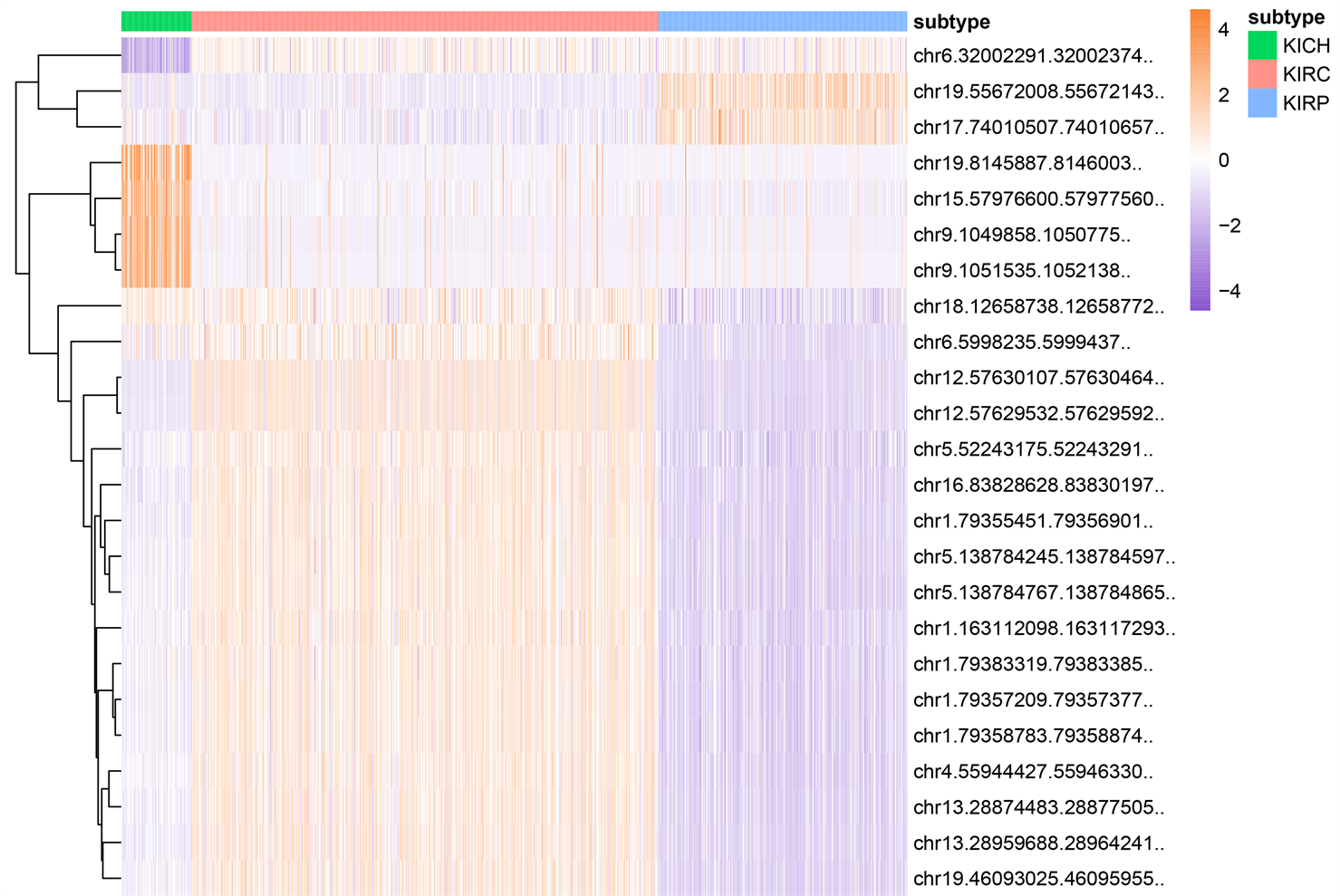
(a) exonRNA-seq重要特征热力图
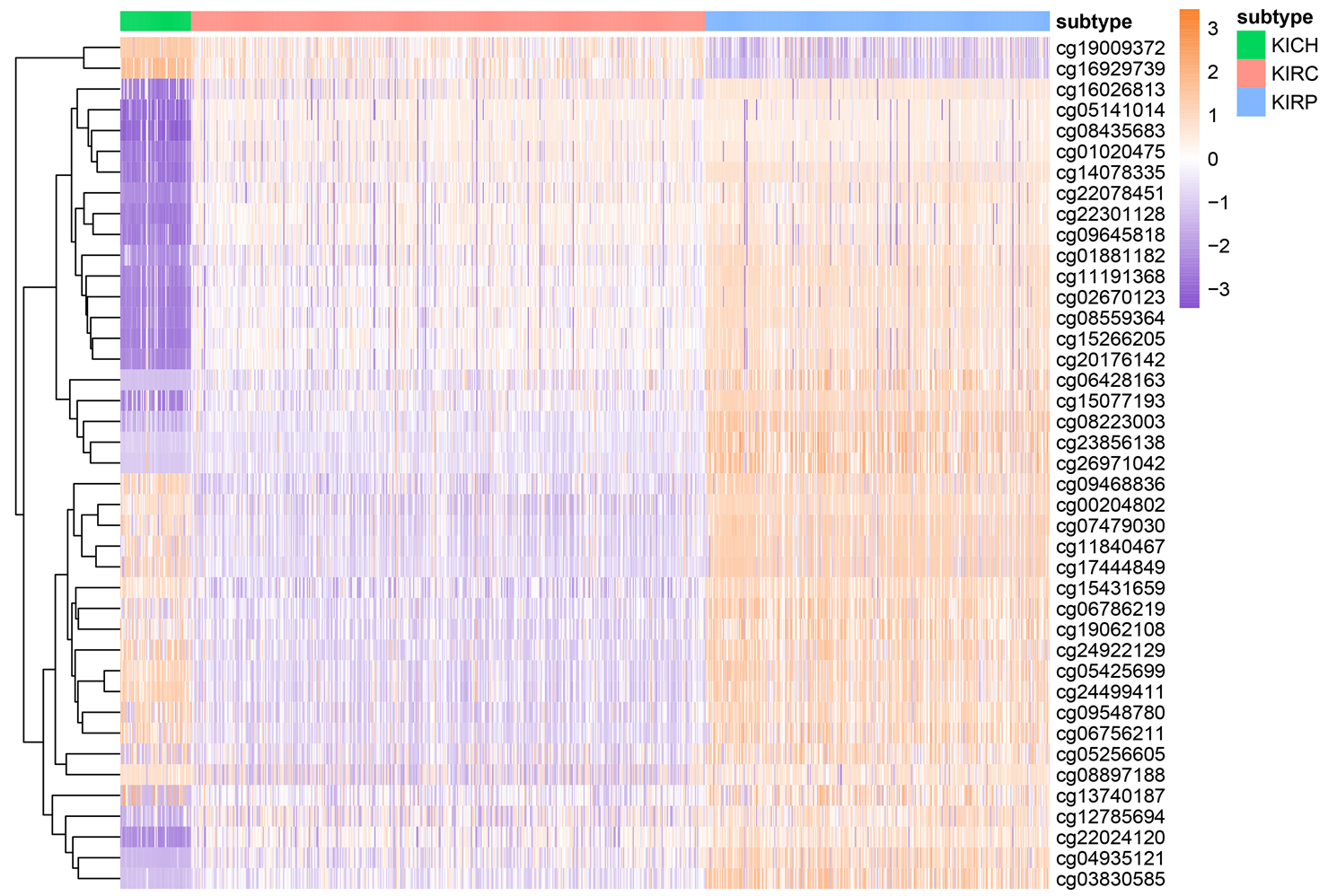
(b) methylation重要特征热力图
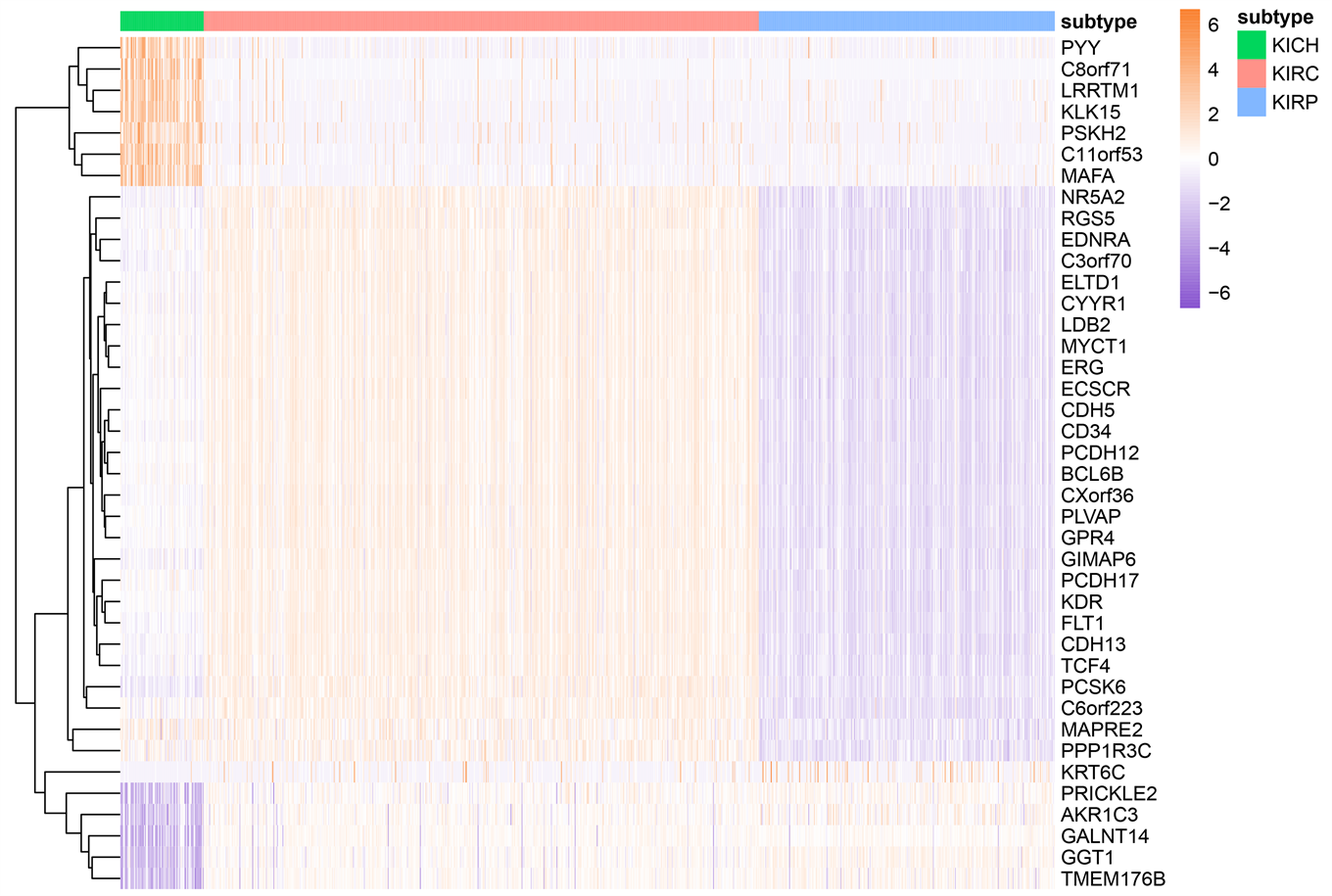 (c) geneRNA-seq重要特征热力图
(c) geneRNA-seq重要特征热力图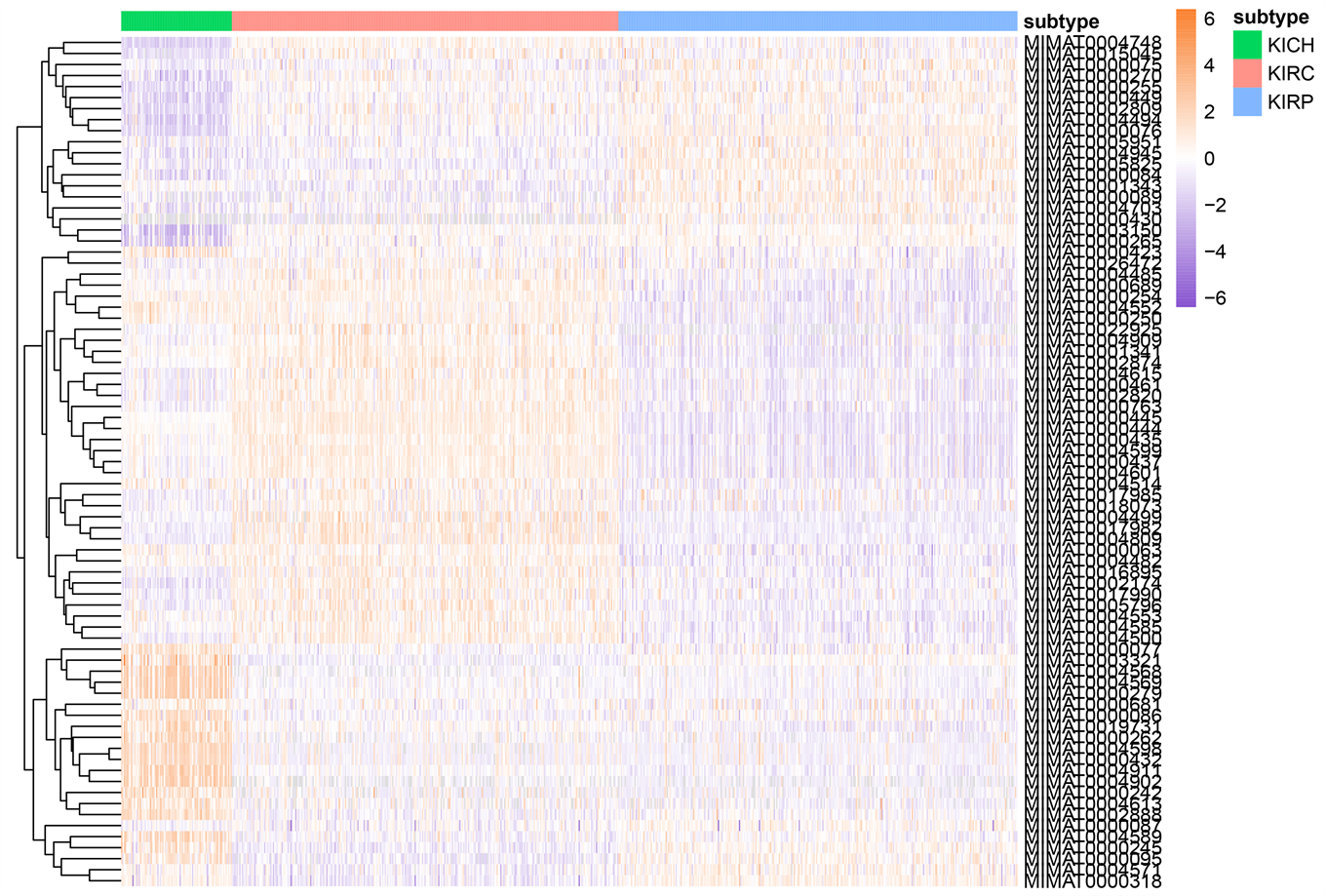 (d) miRNA特征热力图
(d) miRNA特征热力图
Figure S2. Heatmap of selected important features: exon expression level (a), methylation level (b), RNA-seq expression level (c), miRNA mature strand expression level (d)
附图2. 挑选出的重要特征的热力图:外显子的表达水平(a),甲基化水平(b),RNA-seq的表达水平(c),miRNA成熟链表达水平(d)