摘要: 整合多组学数据对癌症患者进行分型,对于提高患者的诊断、治疗和预后效果是至关重要的。传统的统计学方法,例如主成分分析等,对于处理高纬度的多组学数据集的能力有限。为有效整合多组学数据,提出了一种基于卷积神经网络的自编码器框架MCAEI (Multi-Omics Convolutional Autoen-coder Integration)。所提出的卷积自编码器设置了三个卷积层和反卷积层以及一个全连接自编码器来对多组学数据进行压缩和降维,将MCAEI应用于三种癌症并进行了分型工作。此外,所提出的方法与普通、稀疏、降噪自编码器进行比较,实验结果表明MCAEI方法更优。对于得到的最佳生存亚型,还进行了差异基因表达分析和富集通路分析。
Abstract:
Integrating multi-omics data for staging cancer patients is essential to improve patient diagnosis, treatment, and prognosis. However, traditional statistical methods, such as principal component analysis, face limitations when dealing with high-dimensional multi-omics datasets. To effectively integrate multi-omics data, a convolutional neural network-based autoencoder framework, MCAEI (Multi-omics Convolutional Autoencoder Integration), is proposed. The proposed convolutional au-toencoder is composed of three convolutional layers, three corresponding deconvolutional layers, and a fully connected autoencoder. It is utilized to compress and reduce the dimensionality of mul-ti-omics data. The MCAEI method is then applied to three types of cancer for subtype classification. In addition, the proposed method was compared with the normal, sparse, denoising autoencoder. The results demonstrated the superiority of the MCAEI method. For the best survival subtypes ob-tained, differential gene expression analysis and enrichment pathway analysis were also per-formed.
1. 引言
癌症是全球范围内造成死亡人数最多的疾病之一,可以发生在人体的各种组织和器官中。其特点是细胞不受控制的增长,能够通过人体的多个组织传播或增殖,并伴随着细胞层面的分子改变 [1] [2] [3] 。随着高通量测序技术(例如RNA-seq、Methyl-seq、miRNA-seq等)的产生和快速发展,许多研究者致力于从多组学数据的角度来描述癌症的分子和临床特征 [4] 。研究表明,利用多组学数据将癌症患者分为具有一致的分子特征和临床表型的亚组,对于提高患者的诊断、治疗和预后效果是至关重要的 [5] 。癌症基因组图谱(TCGA) (https://www.cancer.gov/types)是一个广泛收集了大量生物学数据的数据库,涵盖了不同的肿瘤类型和基因组平台,这为癌症亚型的分型研究提供了坚实的数据基础。
目前很多方法已被设计,使用多组学数据来识别癌症亚型。在早期,主成分分析和因子分析等传统统计学方法被用来整合多组学数据。例如,iCluster算法 [6] 将肿瘤亚型定义为联合隐变量,基于期望最大化算法(Expectation-Maximization Algorithm)对不同数据类型之间的关联和数据类型内的方差–协方差结构进行建模,并同时减少数据集的维度。Argelaguet等人 [7] 提出了多组学因子分析(MOFA),这是一种基于异构数据的无监督整合方法。它推断出了一种潜在因素,并将其运用到后续的下游分析中。但是主成分分析和因子分析是基于线性假设的降维方法,难以捕获到数据中可能存在的非线性关系。近年来,深度学习(Deep Learning, DL)神经网络已被广泛应用于各个领域 [8] 。它们还对无监督学习产生了积极的影响,学习到的特征可以进一步用于聚类等下游分析任务。而自编码器(Autoencoder, AE)的神经网络结构作为一种数据整合的策略具有广阔的应用前景。AE是一个蝴蝶状的网络结构,其输入层的数据维度与输出层保持一致,并在中间创建一个瓶颈层来提取隐藏特征。其目的是通过学习将高维数据压缩为低维特征表示,并通过最小化损失函数来准确地重构原始数据。Paul等人 [9] 首先根据患者的生存状态对特征进行生存选择,将每个组学中通过检验的特征传递给稀疏自编码器(Sparse Autoencoder, SAE),得到的压缩特征进行谱聚类(Spectral Clustering)分析。为了排除组学数据之间的相互干扰,Wang等人 [10] 提出了一个多模态深度自编码器用来分别学习不同组学数据的高级特征表示,并使用深度神经网络对高级别浆液性卵巢癌的分子亚型进行分类。Liu等人 [11] 使用AE来降低多组学数据维度,融合多组学数据和患者相似度网络建立了一个基于残差图卷积网络(Residual Graph Convolutional Network, GCN)的胃癌分类模型。
本研究结合RNA表达、miRNA表达、拷贝数变异和DNA甲基化四组学数据,提出了一种基于卷积神经网络(Convolutional Neural Network, CNN)的卷积自编码器(Convolutional Autoencoder, CAE)框架MCAEI。采用多组学数据具有更全面地解释复杂的生物学机制和提高分型性能的优势。无论是变分自编码器还是稀疏自编码器,其本质都是在普通自编码器的基础之上引入概率模型或稀疏性约束,可以视为自编码器的特殊形式。而CAE使用了卷积层代替了原来的全连接编码层,卷积层通过在输入数据上滑动滤波器来逐步提取更加抽象的特征表示。相比较于普通自编码器能够更好的处理具有空间结构的数据,并提取更加抽象的特征;具有参数共享、稀疏连接和平移不变性等优点。本文采用一种无监督的学习模式,使用MCAEI框架分别对3种癌症的多组学数据进行特征提取,对具有新特征的样本使用K-means聚类分析,并利用生存分析模型来评估聚类效果。最后,还将我们提出的方法分别与普通、稀疏、降噪自编码器进行比较。结果表明,MCAEI在3个癌症数据集上聚类效果的稳定性更好,能够识别出显著生存差异的癌症亚组,特别是在肾上腺皮质癌数据集上取得了最好的效果。
2. 研究方法
2.1. 数据处理
本文所使用的数据为癌症基因组图谱(TCGA)平台上肾上腺皮质癌(Adrenocortical Cancer, ACC)、乳腺癌(Breast Cancer, BRCA)、肉瘤癌(Sarcoma, SARC)的数据集。每个数据集都包含样本的生存信息(生存状态、生存时间)和四种类型的组学数据(RNA表达、miRNA表达、拷贝数变异和DNA甲基化),见表1。

Table 1. The sample sizes of different omics and survival information data across three types of cancers
表1. 三种类型癌症的不同组学和生存信息数据的样本量
所进行的数据处理过程如下:
1) 将不同组学以及生存信息的样本数据取交集;
2) 删除缺失值比例超过20%的特征数据;
3) 使用Python中的fillna函数填充DNA甲基化特征中的缺失值 [12] 。使用Python软件包scikit-learn中的KNNimpute函数处理miRNA中的缺失值 [13] ;
4) 删除缺失生存数据的样本;
5) 使用一个基因方面的标准差统计量(Gene-Wise Standard Deviation) [14] ,分别选出RNA表达、miRNA表达、拷贝数变异和DNA甲基化中标准差最高的前2200、300、1000、1500个特征;
6) 对所有数据进行最大最小归一化;
7) 根据皮尔逊相关系数对每个组学内的特征进行排序 [15] 。
2.2. 卷积自编码器框架
卷积自编码器是一种基于卷积神经网络(CNN)的结构,通常由输入层、卷积层(Convolutional Layers)、扁平化层(Flatten Layers)、反卷积层(Transposed Convolutional Layers)和输出层组成。本文所提出的MCAEI框架设置了三个卷积层对输入数据进行处理,并将最后一个卷积层扁平化以获取一个高维的长向量;然后,构建了一个具有中间隐藏层的全连接层;最后,通过对完全连接层的输出进行多步反卷积操作,对输入数据进行了重构。具体工作流程如图1所示。

Figure 1. Convolutional autoencoder structure diagram
图1. 积自编码器结构图
一个传统的卷积自编码器由编码器
和解码器
两部分组成。它的目的是通过最小化所有样本数据的输入和输出之间的均方误差(MSE)为每一个输入样本编码。
(1)
对卷积自编码器:
(2)
其中x和h是向量,
是激活函数(例如ReLU型)。在本文中,自编码器堆叠了三个卷积层,并将最后一个卷积层中的单元扁平化形成一个向量。添加了线性整流函数 [16] (Rectified Linear Activation Function, ReLU)到每一层的输出中。
(3)
本文采用了Python语言中的深度学习框架Keras编写了卷积自编码器,并将隐藏层中学习到的压缩特征作为样本的新表示。
3. 实验结果
3.1. 最佳癌症分型
所构建的卷积自编码器的卷积层1、2、3分别设置32、64和128的过滤器;根据自编码器损失函数的变化情况,将训练轮次设置为500轮;优化器采用了Adam优化算法 [17] (Adaptive Movement Estimation Optimization Algorithm);并将学习率设置为0.0005。对所使用的3种癌症数据都进行上述数据处理和分析过程,每一个组学特征通过卷积自编码器被压缩为25个特征,并将四个组学的压缩特征拼接为一个含100特征的矩阵进行聚类。最后,得到癌症分型的轮廓系数(Sil)以及对数秩检验p值,见表2。
Table 2. Comparison of four autoencoders and three types of cancer staging results
表2. 四种自编编码器、三种类型癌症分型结果对比
并且本文还将所提出的卷积自编码器与普通、稀疏、降噪自编码器进行了对比,分别计算了3种癌症对应下的轮廓系数和对数秩检验p值,结果如表2所示。对于普通自编码器将隐藏层的节点数分别设置为500、100、500,输出为一个含100个压缩特征的矩阵,训练批次设置为100。对于稀疏自编码器,模型通过引入稀疏性约束来促使编码器学习到更加稀疏的特征,所以需要更多的训练轮次来达到预期的效果。在本文中,设置训练轮次为1000轮,KL散度惩罚项参数为0.01,学习率0.001。而降噪自编码器设置训练轮次为500轮,噪声系数为0.2。对稀疏与降噪自编码器设置相同的隐藏层节点数100、50、100,输出为一个含50个压缩特征的矩阵。
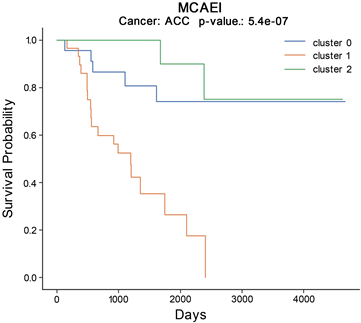
根据分型所得到的轮廓系数和对数秩检验p值结果可以看出,所提出的MCAEI框架在ACC数据集上获得了最好的效果。其中聚类簇数为3时,得到了最低的p值5.3819e-07。由于本文所采用的方法是直接将自编码器所得到的压缩特征进行聚类分析,而不后续再进行COX生存特征选择,这可以实现在缺乏临床生存数据的情况下也能得到很好的分型效果。本文还计算了ACC数据使用普通自编码器并进行COX生存特征选择的情况下,聚类簇数为3时,轮廓系数为0.2360529,p值为4.43744e−05。在BRCA及SARC的数据集上,对于普通自编码器聚类簇数为2时p值很高,并且轮廓系数处于较低范围;对于稀疏自编码器,聚类所得到的轮廓系数虽然较高,但是值均处于较高水平,没有通过对数秩检验;对于降噪自编码器,在SARC数据集上表现不错,得到了较高的轮廓系数和较低的p值,但是在BRCA数据集聚类簇数为2、5时没有通过对数秩检验。本文所提出的卷积自编码器,在不经过COX生存特征选择的情况下,在三个数据集上均获得了较低的p值,并获得了更好的聚类效果。
对于3种癌症,使用本文所提出的框架通过聚类得到最佳的预后亚型。p值最低时,3种癌症的最佳聚类簇数均为3。在最佳聚类簇数下绘制了每种亚型的Kaplan-Meier生存曲线,如图2所示。
 (a) ACC (b) BRCA
(a) ACC (b) BRCA (c) SARC
(c) SARC
Figure 2. Kaplan-Meier survival curve for 3 types of cancer
图2. 种癌症的Kaplan-Meier生存曲线
3.2. 差异表达分析
对于MCAEI框架的差异表达分析,以肾上腺皮质癌ACC为例展开说明。由于对ACC数据进行聚类,簇数为2时获得最高的轮廓系数,故本文将患者数据分为两组(即高风险组与低风险组),使用R语言中的limma软件包进行差异基因表达分析 [18] 。首先对每一个经过方差过滤的基因表达特征,结合癌症分型得到的结果,运用经验贝叶斯方法进行显著性检验。筛选出p < 0.05的所有基因,共383个。并从中选择p值最低的前30个基因绘制差异基因表达热图,如图3所示。
其中C1orf88基因已被证明与ACC的癌症预后有关 [19] 。MSC对形成免疫抑制微环境有重要作用,与ACC患者的生存信息显著相关 [20] 。引入CYP11B1酶的单克隆抗体,能够转化为皮质醇,使得肾上腺皮质结节与其功能之间产生关联 [21] 。由此,通过本文方法得到的分型所识别出的显著基因,能够有效对癌症进行生物学解释。
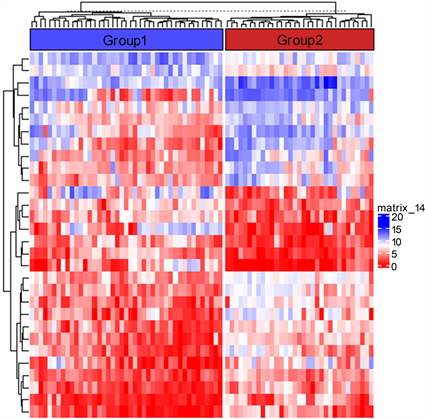
Figure 3. Heat map of differential gene expression for the top 30 genes
图3. 前30个基因的差异基因表达热图
3.3. 富集通路分析
根据得到的差异基因,本文采用R语言中的clusterProfiler软件包进行GO (gene ontology)富集通路分析 [22] 。富集通路分析可以解释一组基因在特定生物过程(BP)、细胞定位(CC)、分子功能(MF)中的富集程度。本文本别计算了这三种基因本体p值最低的前四种富集通路。在BP中,分别是脾脏发育(Spleen Development)、卵泡发育(Ovarian Follicle Development)、造血或淋巴器官的发育(Hematopoietic or Lymphoid Organ Development)、女性性腺发育(Female Gonad Development)。在CC中,分别是中心粒远端下附属物(Centriolar subdistal Appendage)、乙酰胆碱通道复合物(Acetylcholine-Gated Channel Complex)、网络动态蛋白轴丝颗粒(Dynein Axonemal Particle)、磷脂酰肌醇-3-激酶复合物(Phosphatidylinositol 3-Kinase Complex)。在MF中,分别是季氨基结团(Quaternary Ammonium Group Binding)、胞苷酰转移酶活性(Cytidylyltransferase Activity)、乙酰胆碱结合(Acetylcholine Binding)、乙醛脱氢酶(NAD+)活性(Aldehyde Dehydrogenase (NAD+) Activity)。在这些富集通路中,如乙酰胆碱、磷脂酰肌醇-3-激酶等都被证明与肾上腺皮质癌有紧密联系 [23] [24] 。
4. 结语
针对癌症分型方法,本研究提出了一种基于多组学数据和卷积神经网络的自编码器MCAEI框架。该卷积自编码器通过设置了三个卷积层和一个扁平化层来提取对组学数据的压缩特征。所提出的方法能够对3种类型的癌症识别出不同的生存亚型。为了评价本方法的有效性,将所提出的卷积自编码器与普通、稀疏、降噪自编码器进行对比。结果表明,本文所提出的MCAEI框架比其他方法获得了更低的p值和更高的轮廓系数得分。针对癌症分型所得到的结果,本文还进行了差异基因表达分析和富集通路分析,找到了最显著的前30个基因和三种基因本体,这表明了根据本方法的分型结果所识别出的基因,能够进行有效的生物学解释。目前本文的方法仅在3个癌症数据集上进行了测试,未来的工作将使用该方法在更多的癌症数据集上进行测试并对比不同组学数据的效果。