摘要: 朊蛋白病(prion disease)是一类由具传染性的朊蛋白(prion protein, PrP)所致的中枢神经系统变性病。目前已知的人类朊蛋白病主要有克–雅病(Creutzfeldt-Jakob disease, CJD)、格斯特曼综合征(Gerstmann syndrome, GSS)、致死性家族性失眠症(fatal familial insomnia, FFI)、Kuru病。克–雅病(CJD)是最常见的人类朊蛋白病,发病呈全球性分布,发病率约为1/100万。CJD分为散发型、医源型(获得型)、遗传型和变异型四种类型。80%~90%的CJD呈散发型。克–雅病临床早期症状不典型,症状多且异质性大,给该病早期识别和诊断带来了很大困难。现报道1例快速进展的散发型克–雅病病例,总结其临床表现、影像学及相关检验结果、脑电图等,加强对散发型克–雅病的认识。
Abstract:
Prion diseases are a group of degenerative diseases of the central nervous system caused by the infectious prion protein (PrP). The most important known human prion diseases are Creutzfeldt- Jakob disease (CJD), Gerstmann syndrome (GSS), fatal familial insomnia (FFI), and Kuru disease. CJD is the most common human prion disease, with a worldwide distribution and an incidence rate of approximately 0.0001%. It is subdivided into four subtypes: sporadic (sCJD), familial (fCJD), variant (vCJD), and iatrogenic (iCJD). 80%~90% of CJD is sCJD. Due to a variety of factors, including variable presentations and a lack of appropriate gold standard diagnostic methods in the clinical context, it is underdiagnosed and increasingly misdiagnosed. Here, we report a case of sCJD with rapid progression, and summarize its clinical manifestations, imaging and related test results, and electroencephalogram to enhance the understanding of sCJD.
1. 引言
克–雅病(CJD)又称皮质–纹状体–脊髓变性,多隐匿起病,缓慢进行性发展,早期临床表现多变,临床上以进行性痴呆、肌阵挛、锥体束或锥体外系损伤症状为主要表现 [1] 。依据发病病因,CJD分为散发型(sCJD)、医源型、遗传型和变异型,80%~90%的CJD为散发型,sCJD临床早期症状不典型,复杂多变,早期临床诊断困难,现报道1例快速进展的散发型克–雅病病例的临床演变及诊疗过程,以加强临床医生对此病的认识。
2. 临床资料
患者73岁男性,因“言语不清伴左侧肢体无力3天”于2023-10-25入院。患者3天前出现言语不清,能听懂他人言语,伴左侧肢体无力,上肢能抬举但无法持物,不能独立行走,伴头痛头晕,无恶心呕吐,无视物重影,无肢体抽搐及意识障碍,就诊于我院急诊,行颅脑CT示幕上脑白质血管性脱髓鞘可能性大,给予抗血小板,他汀调脂,改善循环等治疗,后患者肢体无力较前好转,为进一步治疗,收住我科。病程中,患者神志清,精神可,饮食睡眠可,体重无明显减轻。既往“胸膜炎”病史,具体治疗不详。“高血压”病史,未服用降压药物。“肋骨骨折病史”,未特殊治疗。个人史及家族史无特殊。
入院神经系统查体:神志清楚,言语欠流利,地点定向力下降,人物、时间定向力正常,眼动可,无眼震,双侧瞳孔等大等圆,直径3 mm,对光反应可,左侧鼻唇沟浅,伸舌偏左,右侧肢体肌力5级,左上肢肌力4级,左下肢肌力4级,肌张力正常,左侧偏身痛温觉减退,左侧巴氏征阳性,左侧指鼻及跟膝胫试验欠稳准,脑膜刺激征(−)。
辅助检查:2023年10月23日颅脑CT平扫:幕上脑白质血管性脱髓鞘可能性大,老年性脑改变。胸部CT平扫:右肺陈旧性结核可能性大,左肺少许小结节,双肺慢性炎症,双肺气肿,肝多发囊肿。胸部(肋骨) CT三维成像:右侧第7肋骨后段、左侧第9肋骨后段、左侧第4肋骨前段、右侧第8肋骨后段骨折。
诊疗经过:患者入院后继续给予抗血小板、调脂、改善循环治疗,继续完善相关检验检查,降钙素原检测、CK + CKMB测定、电解质检测、血同型半胱氨酸测定、转氨酶测定(ALT/AST)、白蛋白测定、肾内科肾功、尿酸测定、总胆固醇测定、甘油三酯测定、低密度脂蛋白胆固醇测定、肝功、血氨测定、传染性标志物4项检测、尿液分析、粪便常规分析 + 隐血试验、抗中性粒细胞胞浆抗体测定、ENA抗体谱、抗磷脂抗体测定等未见明显异常。血常规(无网红) + CRP:白细胞计数14.66 × 109/L,中性粒细胞计数9.92 × 109/L,淋巴细胞计数3.28 × 109/L,单核细胞计数1.12 × 109/L,血小板392 × 109/L,血小板比积0.40%,全血C反应蛋白28.63 mg/L;血凝常规:纤维蛋白原5.99 g/L,D-二聚体640.00 ng/mL;空腹血糖:6.39 mmol/L;血沉检测:红细胞沉降率30.00 mm/h;男性肿瘤标志物筛查:神经元特异性烯醇化酶22.90 ng/mL,糖类抗原724 7.38 U/mL。2023年10月25日完善颅脑MR示脑白质血管源性脱髓鞘改变。2023年10月27日完善常规脑电示右侧前头部(额极、前中颞区)为主中–高波幅慢波,持续发放,间少量尖波(见图1)。
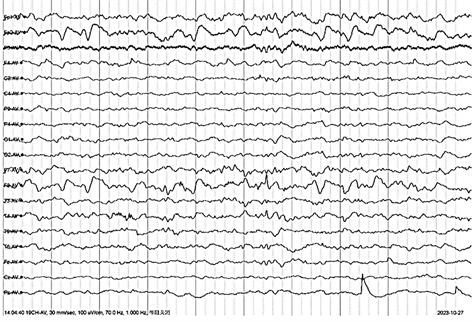 注:右侧前头部(额极、前中颞区)为主中–高波幅慢波,持续发放,间少量尖波。
注:右侧前头部(额极、前中颞区)为主中–高波幅慢波,持续发放,间少量尖波。
Figure 1. 2023-10-27 EEG result
图1. 2023-10-27 EEG检查结果
于10月28日患者出现精神烦躁,言语混乱,不认识家人,伴有发作性肢体抖动,每次持续几秒钟,10月30日患者病情进一步进展,患者嗜睡,精神较差,伴有发作性烦躁,不思饮食,无法与人正常交流,无发热,无意识丧失。体格检查:嗜睡状态,不言语,查体不合作,双侧瞳孔等大等圆,直径3 mm,对光反应迟钝,左侧鼻唇沟浅,四肢肌张力增高,有少许自发活动,左侧巴氏征阳性,左侧踝振挛阳性。进一步完善腰椎穿刺并行脑脊液检查,并外送脑脊液及血清自身免疫性脑炎相关指标,结果示腰穿压力:110 mm H20,脑脊液免疫球蛋白测定:脑脊液IgA 5.66 mg/L,脑脊液IgG 38.20 mg/L。脑脊液细菌涂片三项检测、脑脊液常规检查、脑脊液生化未见明显异常。复查颅脑MR平扫显示双侧额顶岛颞叶皮层DWI信号增高(见图2)。
患者诊断不明,病情进展迅速,主要表现为认知功能下降和精神症状为著,伴有发作性肢体抖动,结合患者病史、临床表现及现已经回示的相关辅助检查,考虑诊断:自身免疫性脑炎?克雅氏病?线粒体脑病?病毒性脑炎?告知家属病情后,酌情加用免疫球蛋白及激素治疗,病毒性脑炎不除外,给予抗病毒药物治疗。经上述治疗后,患者症状出现短暂性改善,右上肢可配合指令抬举。患者脑脊液及血清自身免疫性脑炎相关抗体检测结果回示阴性。后续患者再次出现病情进展,出现惊恐发作,11月10日再次行24小时视频脑电监测,示背景节律减慢,右侧额极、额、前中颞区尖慢复合波、慢波近持续发放,部分呈周期样发放(见图3),于11.13再次行3小时视频脑电,开始时脑电监测可见右侧额极、额、前中颞区尖慢复合波呈周期或类周期样持续发放(见图4),给予安定10 mg缓慢静推,开始静推约2分钟,右侧额极、额、前中颞区尖慢复合波消失,背景可见8~9 Hz低–中波幅α节律(见图5),后续给予安定2支入液持续泵入,但约30 min分钟后恢复至用药前脑电图状态,后续持续给予安定小剂量静脉泵入,患者症状仍未见好转,排除非惊厥性癫痫持续状态。于11月15日再次完善腰穿,并外送脑脊液DNA + RNA病原学检测(NGS)及14-3-3,结果回示,NGS阴性,脑脊液14-3-3蛋白阳性。综合患者入院后检验检查及相关治疗,综合考虑为很可能CJD。后续患者病情进一步进展,发展至无动性缄默–木僵状态,后患者家属自动出院。
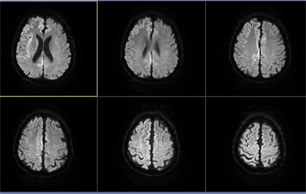
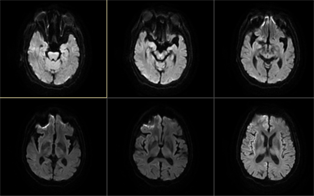 注:双侧额顶岛颞叶皮层DWI信号增高。
注:双侧额顶岛颞叶皮层DWI信号增高。
Figure 2. Results of DWI examination of the brain
图2. 颅脑DWI检查结果
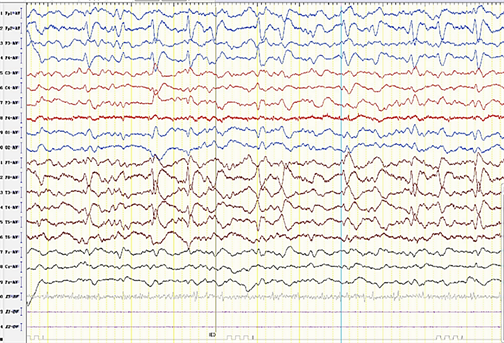 注:右侧额极、额、前中颞区高波幅尖慢复合波、慢波近持续发放,部分呈周期样发放。
注:右侧额极、额、前中颞区高波幅尖慢复合波、慢波近持续发放,部分呈周期样发放。
Figure 3. 2023-11-10 EEG result
图3. 2023-11-10 EEG检查结果
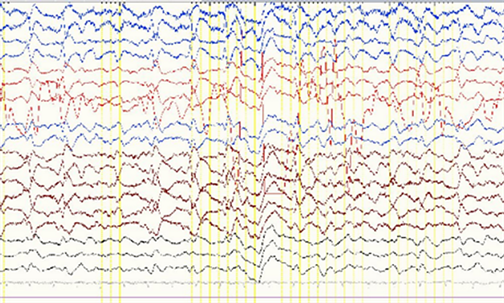 注:右侧额极、额、前中颞区尖慢复合波呈周期或类周期样持续发放。
注:右侧额极、额、前中颞区尖慢复合波呈周期或类周期样持续发放。
Figure 4. 2023-11-13 EEG result before sedation with Valium
图4. 2023-11-13静推安定之前脑电监测结果
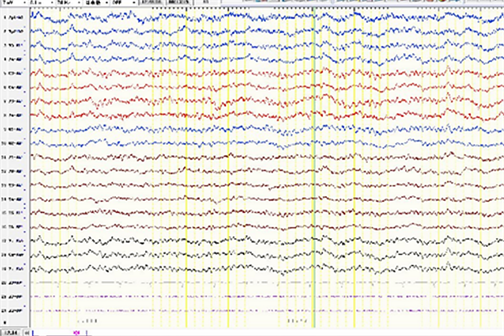 注:右侧额极、额、前中颞区尖慢复合波消失,背景可见8~9 Hz低–中波幅α节律。
注:右侧额极、额、前中颞区尖慢复合波消失,背景可见8~9 Hz低–中波幅α节律。
Figure 5. 2023-11-13 EEG result 2 minutes after sedation with Valium
图5. 2023-11-13静推安定2分钟之后脑电监测结果
3. 病例讨论
克雅氏病发病年龄为25~78岁,平均58岁,男女均可罹患。患者多隐匿起病,缓慢进行性发展。典型的CJD临床可分为三期 [2] 。初期表现为易疲劳、注意力不集中、失眠、抑郁和记忆力减退等症状;中期表现为大脑皮质、锥体外系、锥体束及小脑受损的症状;晚期出现无动性缄默、去皮质强直或屈曲性四肢瘫痪伴挛缩,多因压疮或者肺部感染死亡 [1] 。本例患者以言语不清伴左侧肢体活动不灵起病,起初考虑为脑血管病,但完善影像学检查未见出血及新发梗死灶,且病情进展迅速,患者在起病两周内迅速发展至晚期,发展至无动性缄默–木僵状态,追问家属,在此次起病之前患者无手术、血液制品接触史,近期无生食、旅游史,患者起病前无进行性记忆力下降等痴呆类症状,在此期间我们继续完善脑电图、影像学、腰穿等检验检查,找到了更多支持朊蛋白病的可能性,为临床诊断克雅病提供了更多参考。
克雅病的诊断可采用以下标准 [3] :① 在2年内发生的进行性痴呆;② 肌阵挛、视力障碍、小脑症状、无动性缄默等四项中具有其中两项;③ 脑电图显示周期性同步放电的特征性改变。患者具备以上三项可诊断很可能;仅具备①②两项,不具备③项诊断为可能CJD;如患者脑活检发现海绵状态和PrPsc为确诊的CJD。现阶段克雅病的确诊依赖于脑组织病理检测,显著的组织病理改变为脑组织呈海绵状病变 [4] ,但因脑组织活检创伤及手术风险高,对手术者操作要求高,患者及其家属较难接受,故本例患者未接受此检查。
头颅影像学是目前诊断克雅病的主要辅助检查之一。头颅MRI中弥散加权像(DWI)序列在克雅病诊断中具有重要价值,典型表现为异常高信号呈对称性或非对称性分布于大脑皮层和基底节区,主要见于额叶、颞叶等部位,又称为“缎带征” [5] ,部分研究发现,CJD的头颅磁共振异常信号区域无强化表现,这也是CJD的特征性表现之一 [6] 。sCJD患者病例中均存在灰质高信号(DWI > FLAIR),某些区域优先受累,但是并不局限在边缘区域,所有具有基底神经节或丘脑DWI高强度的sCJD都有相关扩散受限,但是在非朊病毒引起的例如自身免疫性脑炎、单纯疱疹病毒性脑炎、亨廷顿病中均未发现这种灰质高信号限制的扩散 [7] 。少数病人皮层DWI阳性,而ADC序列为阴性,可能是因为涡电流失真现象或肉眼分析ADC影像辨别力差 [8] 。本例患者头颅MRI中DWI显示对称性或不对称性皮质“缎带征”,增强MR未见异常信号区域强化,这与克雅病颅脑MRI表现相符合。
散发型克雅病的诊断标准,其辅助检查还主要参考脑脊液14-3-3蛋白或脑电图示周期性尖–慢复合波 [9] 。免疫荧光检测脑脊液中14-3-3蛋白可呈阳性,对克雅病的诊断具有重要价值。脑脊液14-3-3蛋白在正常脑组织中含量丰富,当感染了传染性朊蛋白后,大量脑组织被破坏,蛋白漏于脑脊液中,诊断很可能散发型克雅病的敏感性为90%~97%,特异性为87%~100% [10] 。在其他疾病如于阿尔茨海默病、中枢神经系统感染、桥本脑病等可导致神经细胞变形坏死的疾病,也可见脑脊液14-3-3蛋白阳性。在本例病例中患者脑脊液14-3-3外送结果为阳性,为散发型克雅病进一步提供了证据。脑电图周期性同步放电被认为是CJD的特征性改变 [11] ,但散发型CJD患者的脑电图表现随着病程的演进而发生改变 [12] [13] 。早期患者的脑电波慢波活动增多,基本节律解体;中晚期患者的脑电图是以θ波、δ波为主的弥漫性、广泛性慢波,随病情加重出现广泛同步的周期性尖慢复合波,即尖波(双相或三相)、复合波(由棘波、多棘波及慢波组成),时程100~600 ms,发放周期为0.5~2.0 s,具有诊断意义,末期患者的脑电波周期间隔不变或延长,尖波波幅逐渐消失 [14] [15] 。本例患者第一次完善脑电检测可见持续发放的中–高波幅慢波,后续患者病情急剧恶化,再次完善脑电图提示周期性发放的尖慢复合波,后续患者自动出院,未监测到晚期脑电表现。结合患者出现肌阵挛表现,故再次支持了sCJD的诊断。虽然老年性痴呆及路易体痴呆也会有类似的脑电图表现,但老年性痴呆病程相对长,路易体痴呆症状往往有波动,故对于患有快速进行性痴呆和局灶性神经系统体征的患者,且合并周期性尖慢复合波的脑电图特点,CJD应该是一线诊断 [16] 。
4. 结论
综上所述,sCJD是由PrP感染的快速进展的致命的神经变性病,当临床表现为快速进展的认知功能障碍、肌阵挛、锥体束/锥体外系损害、小脑/视觉异常等时,应行颅脑MRI、EEG、脑脊液14-3-3蛋白查以明确临床诊断。本病例在发病早期以脑血管病症状入院,但完善影像学检查未见相关脑血管病影像学证据,后期患者出现意识及肌张力变化,且此病例病情进展迅速,患者入院时神志清楚,两周内迅速发展至无动性缄默–木僵状态,这为在以后的临床工作中提供了宝贵经验。患者急性起病,快速出现认知障碍,头颅MRI病灶不符合脑血管分布特点时,不应考虑脑血管病;在脑脊液常规、生化、脑脊液细菌培养、自身免疫性脑炎相关抗体检查中均为阴性,且经使用激素冲击、抗病毒、改善循环等治疗后患者病情未见好转,仍呈进展趋势时不应考虑为脑炎或脑血管病;头颅磁共振未见脑实质局灶性病变,而表现为双侧基底节区、皮层异常信号,这个时候应考虑为CJD的可能;对于高级神经功能下降的患者应及时完善脑电图检查,临床上出现痴呆、肌阵挛、周期性脑电异常放电的老年患者常提示CJD [17] 。
sCJD目前尚无有效治疗方法,预后极差,只有尽早诊断,早期采取对症治疗,以延长患者生存期。