摘要: 横纹肌肉瘤作为一种起源于横纹肌细胞或有倾向分化为横纹肌细胞的间叶细胞的恶行肿瘤,由各种不同分化程度的横纹肌母细胞组成,是儿童软组织肉瘤中最常见的一种。横纹肌肉瘤发病率次于恶性纤维组织细胞瘤和脂肪肉瘤,按组织类型可分为胚胎性横纹肌肉瘤、腺泡状横纹肌肉瘤及多形性横纹肌肉瘤,其中胚胎性横纹肌肉瘤(ERMS)约占横纹肌肉瘤的2/3,好发于儿童及青少年,尤其是15岁以下的儿童。多项研究表明,横纹肌肉瘤生存率5年总体生存率(OS)位于70%~90%之间,5年无事件生存率(EFS)位于60%~80%,但长期预后仍欠佳。本文针对1例儿童发病、发生于宫颈的胚胎性横纹肌肉瘤进行临床诊疗相关病例汇报,并复习相关文献。
Abstract:
Rhabdomyosarcoma, as a malignant tumor originating from striated muscle cells or mesenchymal cells that tend to differentiate into striated muscle cells, is composed of various degrees of differen-tiation of striated myoblasts and is the most common type of soft tissue sarcoma in children. The in-cidence rate of rhabdomyosarcoma is second to that of malignant fibrous histiocytoma and liposar-coma. It can be divided into embryonic rhabdomyosarcoma, alveolar rhabdomyosarcoma and pol-ymorphic rhabdomyosarcoma according to the tissue type. Among them, embryonic rhabdomyo-sarcoma (ERMS) accounts for about 2/3 of rhabdomyosarcoma, which is most common in children and adolescents, especially children under 15 years old. Multiple studies have shown that the over-all 5-year survival rate (OS) of rhabdomyosarcoma is between 70% and 90%, and the 5-year event free survival rate (EFS) is between 60% and 80%, but the long-term prognosis is still poor. This ar-ticle reports a clinical diagnosis and treatment related case of embryonic rhabdomyosarcoma of the cervix in a child, and reviews relevant literature.
1. 病例汇报
患者女性,13岁,无性生活史,因“阴道口肿物脱出3月”于2022-08-02收入院。患者于2022年5月出现阴道口肿物脱出,偶有脱出,脱出大小约0.5 cm,无触痛,6月于我院查超声未见明显异常,7月21日月经来潮,阴道口脱出肿物较前明显增大,脱出物大小约2 cm,无触痛。7月29日我院就诊查妇科超声提示:阴道回声较杂乱,内见数个囊性回声及点状强回声,囊性回声较大者约1.5 × 0.6 cm,透声可。阴道内囊性回声,考虑囊肿可能,点状回声考虑钙化可能。妇科查体:外阴发育正常,肛诊未扪及明显异常。遂行宫腔镜下宫颈管息肉电切术 + 宫腔镜检查,术中见:阴道内见约4 × 4 × 3 cm分叶状质软息肉样物,有一细蒂连于宫颈管中段前壁,蒂粗约0.3 cm,血运稍丰富,未再探查宫腔。术后病理检查:大体观察息肉样组织一块,大小4 × 3 × 1.5 cm,切面质软胶冻样。病理诊断为恶性肿瘤,意见为肉瘤,考虑胚胎性横纹肌肉瘤可能,后加做免疫组化示CK (灶+),Vimentin (+),Desmin (部分+),Myogenin (部分+),MyoD1 (部分+),Ki-67 (+,约80%),ALK (5A4) (−),WT-1 (灶+),意见为胚胎性横纹肌肉瘤,部分区域肿瘤细胞异型明显,见瘤巨细胞,呈间变性改变,核分裂像多见(约20个/10HPF)。目前行VAC方案(长春地辛 + 吡柔比星 + 卡铂)化疗中。
2. 讨论
横纹肌肉瘤按组织类型可分为胚胎性横纹肌肉瘤、腺泡状横纹肌肉瘤及多形性横纹肌肉瘤 [1] ,其中低、中、高危组及中枢侵犯组OS率及EFS率依次递减 [2] [3] ,胚胎型横纹肌肉瘤好发于头部、颈部、泌尿生殖道及腹膜后,主要表现为痛性或无痛性肿块 [4] ,生长较快。目前尚未发现特异性的实验室指标辅助诊断,影像学检查一般用于判断原发灶大小、组织浸润及骨受累情况,但其影像学特征往往只具备恶性肿瘤共性,缺少特异性,需结合临床表现及患者年龄性别等综合诊断,在后期治疗过程中常常用于评估治疗反应 [5] [6] ,确诊的唯一“金标准”是病理组织学诊断 [7] 。
根据国际儿童肿瘤研究协会指定的临床分期系统(见图1)及美国横纹肌肉瘤研究(IRS)组的术后–病理临床分组系统(见图2)可将胚胎横纹肌肉瘤分为低危组、中危组、高危组、中枢侵犯组(见表1)。

Figure 1. Clinical staging system (from the Internet)
图1. 临床分期系统(来自网络)
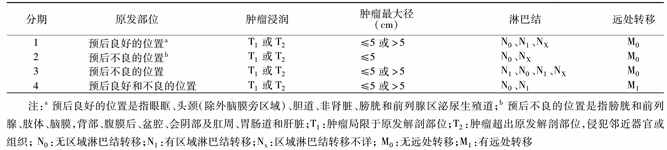
Figure 2. Postoperative-pathological clinical grouping system for IRS (from the Internet)
图2. IRS的术后–病理临床分组系统(来自网络)

Table 1. Risk grouping of embryonal rhabdomyosarcoma
表1. 胚胎性横纹肌肉瘤危险程度分组
注:中枢侵犯组指同时伴有颅内转移扩散、脑脊液阳性、颅底侵犯或者颅神经麻痹中任意一项。
手术治疗是ERMS主要的治疗方法,一般行广泛性局部切除术,需切除肿瘤及其周围的部分组织,尽可能的切除可见的病灶,做到完整切除或切除后仅有镜下残留对于预后来说至关重要 [8] ,多个单中心研究表明局部治疗的欠缺可能是导致复发或进展的重要因素,对肿瘤切缘情况进行生存分析,发现术中肉眼残留和镜下残留患儿存活率明显低于无残留患儿存活率 [9] [10] ,ERMS的IRS I期不行放疗,而II~IV期则需要行放疗。一般术后7天内开始化疗,通常选用VAC方案,根据不同危险分级选择不同强度方案,完全缓解后4~6疗程可考虑停药,超过12个疗程可根据个体进行调整 [11] 。
基因检测对于患者的诊断及后续方案的制订与预后均有着密切的联系,它与年龄、肿瘤部分、组织学分型、手术切除完整程度及肿瘤扩散程度相结合,可将患者进行分层。胚胎性横纹肌肉瘤存在多种遗传改变,其主要特征为PAX-FOXO1融合阴性,且伴有复杂的基因组改变,如染色体8、2、11、12、13和/或20增加和10和15丢失,11p染色体上的杂合性严重丧失 [12] 。而最新研究针对TP53突变进行分析,认为在融合阴性和融合阳性的病例中,TP53突变提示不良的预后 [13] 。
除此之外,免疫治疗的研究也在不断展开。最新研究根据47例融合基因阴性RMS及44例融合基因阳性患者的总生存率制作了生存曲线图,图像显示分子特征显示融合阴性预后较融合阳性好,二者存在分子亚型间及分子亚型内细胞成分及分化状态差异,融合阴性内存在类似正常肌细胞分化状态,而分化程度较高细胞比例越高预后越好 [14] 。基于横纹肌肉瘤的分子特征及免疫组化等研究,免疫治疗或可成为治疗横纹肌肉瘤另一值得探寻的领域。
目前针对于横纹肌肉瘤的探讨仍局限于小样本量单中心研究,希望能开展大样本量多中心的研究,为横纹肌肉瘤的诊治提供更多数据支持。