摘要: 目的:分析Gitelman综合征的临床诊断要点和治疗随访经验,为Gitelman综合征患者的早期诊断、规范治疗提供思路。方法:选取1例因生长迟缓就诊的Gitelman综合征患儿作为研究对象,收集患者的临床资料,通过全外显子组测序筛选候选变异,并进行Sanger测序验证,随访患者的治疗效果。结果:本例13岁患儿因生长迟缓就诊,实验室检查示低钾低镁血症、高肾素、肾性失钾,基因检测发现SLC12A3基因复合杂合变异,确诊为Gitelman综合征,予规范治疗并随访1年2个月,患儿身高、体重明显增长,心电图的ST段压低恢复,生活质量显著改善。其中父源的c.791delinsGCGTGGTCTCGGTCATT GG单条变异为新发现的基因突变位点,丰富了Gitelman综合征的基因突变谱。结论:因生长迟缓就诊的患儿,需重视鉴别诊断,对于低钾血症尤其合并低镁血症、肾性失钾的患儿需警惕Gitelman综合征,及早诊治、规范管理以达最佳预后。
Abstract:
Objective: To analyze the clinical diagnostic points, treatment and follow-up experience of Gitelman syndrome, to provide ideas for the early diagnosis, standardized treatment of Gitelman syndrome patients. Methods: A child with Gitelman syndrome who was treated for growth retardation was selected as the study object, and his clinical data was collected. Candidate variant was identified through whole exome sequencing, and Sanger sequencing was used for validation. The therapeutic effect of the patient was followed up. Results: In this case, a 13-year-old patient was treated for growth retardation and laboratory examination showed hypokalemia, hypomagnesemia, hyper-renin and renal potassium loss. Genetic test found compound heterozygous variation of SLC12A3 gene, and the patient was diagnosed with Gitelman syndrome. After receiving standardized treatment and follow-up for 1 year and 2 months, the patient’s height increased significantly, the ST segment depression of the electrocardiogram recovered, and the quality of life improved significantly. The single variant of c.791delinsGCGTGGTCTCGGTCATT GG from the father is a newly discovered gene mutation site, enriching the gene mutation spectrum of Gitelman syndrome. Conclusion: Children with growth retardation should pay attention to differential diagnosis. For children with hypokalemia, especially those with hypomagnesemia and renal potassium loss, it is necessary to be vigilant about Gitelman syndrome, so as to achieve the best prognosis through early diagnosis and treatment and standardized management.
1. 引言
Gitelman综合征(Gitelman Syndrome, GS, OMIM263800),又称家族性低钾低镁血症,是一种罕见的常染色体隐性遗传的肾小管疾病,以低钾血症、低镁血症、低氯血症、低钙尿症、偏低血压和高肾素活性等“五低一高”为主要临床特征 [1] 。欧洲人中GS的患病率约为(1-10)/40,000,亚洲人群中可能更高 [2] 。GS患者起病较晚,通常在青春期或成年期,临床表现具有高度异质性,多数患者症状较轻,仅表现为低钾血症引起的肌无力、乏力、肌肉痉挛等,部分患者仅生化指标异常,无明显临床症状,甚至缺乏典型的生化异常,易被误诊、漏诊 [3] 。GS患者的生活质量远低于正常人群,早期诊断和治疗对GS患者的预后具有重要意义。现回顾性分析一例因生长迟缓就诊并诊断为GS的青少年患者的临床资料,并复习国内外相关文献,为GS的临床早期诊治、规范管理提供依据。
1.1. 病例资料
患儿,男,13岁8月,因“自幼身材较同龄儿矮小”就诊,患儿自幼生长速度缓慢,身高增长约3~4 cm/年,平素懒动、易疲劳,饮食睡眠尚可,二便正常。既往无特殊病史。父亲178 cm,母亲158 cm,16岁姐姐158 cm,均体健,父母非近亲结婚,无家族遗传性疾病,患儿系G2P2,足月剖宫产,出生体重3.6 Kg,身长50 cm,出生史无异常。体格检查:W 29.7 Kg (−2.99 SD),H 143.0 cm (−2.92 SD),BMI:14.5 kg/m2,BP:100/60 mmHg (1 mmHg = 0.133 kPa)。一般情况可,身材匀称,面色欠红润,双肺呼吸音清晰,未闻及干湿性啰音,心律齐,心音有力,各瓣膜听诊区未闻及杂音,腹软,肝脾肋下未触及,四肢肌力、肌张力正常。外阴发育Tanner2期,睾丸容量约6 ml。初步诊断为身材矮小症,为进一步查找病因,完善相关检查:骨龄片示11岁左右,心电图示窦性心律不齐、电轴左偏、ST段改变,血常规、尿常规、肝肾功、甲功三项、空腹血糖、血脂分析基本正常,电解质分析示:血镁:0.57 mmol/L (0.75~1.02 mmol/L),钾:3.39 mmol/L (3.5~5.3 mmol/L),血氯:98.70 mmol/L (99~110 mmol/L)。因身材矮小、骨龄落后,查胰岛素样生长因子(insulin-like growth factor, IGF) 135 ng/ml (<−2 SD),生长激素激发试验示GH峰值4.04 ng/ml,垂体核磁共振检查未见异常。结果提示患儿低钾、低镁、低氯血症,同时存在生长激素缺乏。为进一步寻找电解质异常的原因,行24小时尿电解质检测:NA/24H:100.83 mmol/24H,CL/24H:119.42 mmol/24H,K/24H:36.03 mmol/24H,肾素:11.69 ng/(mL∙hr) [0.10~6.56 ng/(mL∙hr)]、醛固酮、血管紧张素、促肾上腺皮质激素、皮质醇正常,泌尿系超声检查未见异常,结果提示患儿尿钾排泄量增加,符合肾性失钾,同时肾素–血管紧张素–醛固酮系统(Renin angiotensin aldosterone system, RAAS)激活。
1.2. 基因检测
患儿生长迟缓、乏力、低血钾、低血镁、低血氯合并高肾素、肾性失钾(血清钾 < 3.5 mmol/L时,尿钾排泄量 > 25 mmol/24H),临床疑诊GS,遂经患儿父母同意后采集患儿和父母及其姐姐外周血送北京迈基诺医学检验所,提取基因组DNA,捕获外显子,对患儿DNA行全外显子组基因检测,发现患儿SLC12A3基因复合杂合突变:c.791delinsGCGTGGTCTCGGTCATT GG (p.A264delinsGVVSVIG)和c.179C > T (p.Thr60Met),经家系验证分析,患儿父母分别携带单条变异,患儿姐姐无此基因突变,见图1。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)指南,两变异判定为疑似致病性变异(Likely pathogenic)。其中,整码突变c.791delinsGCGTGGTCTCGGTCATTGG (p.A264delinsGVVSVIG)为文献数据库尚未报道的突变,为新发现的SLC12A3基因突变位点。
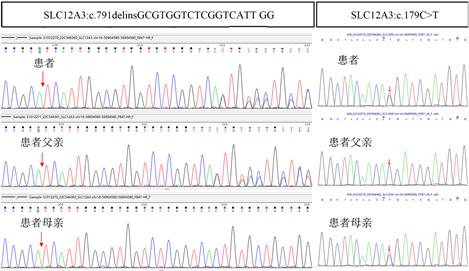 注:变异c.791delinsGCGTGGTCTCGGTCATT GG、c.179C > T分别遗传自父亲、母亲。
注:变异c.791delinsGCGTGGTCTCGGTCATT GG、c.179C > T分别遗传自父亲、母亲。
Figure 1. SLC12A3 gene sequencing results
图1. SLC12A3基因测序结果
1.3. 诊断
参照《Gitelman综合征诊治中国专家共识(2021版)》临床诊断标准,根据患儿临床表现、实验室检查和高通量测序结果,患者确诊为GS,合并生长激素缺乏。
1.4. 治疗和随访
指导患儿多食用富含钾、镁的食物,同时口服门冬氨酸钾镁片、维生素D、钙镁片,给予重组人生长激素(Recombinant human growth hormone, rhGH) 4u皮下注射Qn [0.13 u/(kg∙d)],定期测量身高、体重并绘制生长曲线(图2),可见患儿身高、体重增长明显。定期复查患儿甲状腺功能、电解质指标和心电图,患儿血钾、血镁维持在Gitelman综合征控制目标以上,即血钾 > 3.0 mmol/L,血镁 > 0.5 mmol/L [4] (表1)。患儿心电图的ST段压低已改善(图3)。尤为重要的是,患儿目前不再感觉乏力,运动能力大幅提高,生活质量得到明显改善,这与我们的治疗目标相符,继续动态随访患儿上述指标变化。

Figure 2. Changes in Z-scores of height and weight in pediatric patients before and after treatment
图2. 患儿治疗前、后身高及体重的Z分值变化

Table 1. Comparison of biochemical indicators before and after treatment in pediatric patients
表1. 患儿治疗前、后生化指标对比

Figure 3. Changes in electrocardiograms of pediatric patients before and 10 months after treatment
图3. 患儿治疗前、治疗10个月后心电图对比
2. 讨论
GS的临床确诊依赖于基因检测,编码噻嗪类利尿剂敏感的Na+/Cl−共同转运体的SLC12A3双等位基因失活突变是确诊Gitelman综合征的金标准 [5] 。随着近年来对Gitelman综合征研究的深入及基因检测的普及,越来越多的研究显示编码氯通道ClC-Kb的CLCNKB基因突变可能也是GS的致病基因,增加了基因诊断的难度及复杂性 [6] 。到目前为止,已报道的SLC12A3基因突变超过500个,70%以上的GS患者为复合杂合突变 [7] 。GS患者的临床表型与基因型之间相关性尚无明确定论,RIVEIRA-MUNOZ E等人对27名诊断为GS的患者的基因型及临床表型进行相关性分析,并以动物实验表明SLC12A3突变的性质/位置以及男性性别是GS患者严重表型的决定因素 [8] 。而LEE等人对34名患者进行队列研究表明性别、基因型或SLC12A3突变等位基因的数量不能预测疾病的严重程度或对治疗的反应 [9] 。GS的临床问题多与电解质紊乱及RAAS激活等有关,可累及全身多系统,早期一般表现为低钾所致的乏力、嗜盐、多尿等非特异性症状,随着病程的延长,长期的低钾低镁可致肾功能损害、糖代谢异常、生长发育迟缓等严重并发症 [4] 。因此早期诊断、早期治疗非常必要。既往研究表明,GS患者不同形式的治疗虽未能完全纠正低钾、低镁血症,但可显著改善身材矮小患者的生长速度和IGF-I水平,改善相关临床症状 [10] 。
生长迟缓和身材矮小是常见的儿科问题,BACKELJAUW P等人使用Embase、MEDLINE和Cochrane数据库检索2008至2020年期间有关身材矮小对成人和儿童造成的负担的研究,有证据表明,与正常身材的人相比,任何原因导致身材矮小的儿童和成年人会经历更差的生活质量 [11] 。引起矮小的病因繁多,通过详细的病史询问、系统的体格检查、基本的实验室及必要的影像学检查可以除外部分内分泌疾病、营养问题及慢性全身性疾病,遗传性疾病所致的矮小需要完善染色体核型分析或基因检测以明确诊断。本例患者因生长迟缓就诊,实验室检查提示生长激素完全缺乏,如果不经过缜密的鉴别诊断,并进行针对性的基因检测,很容易误诊为“生长激素缺乏症”,直接予以生长激素补充治疗。虽然GS患者合并生长激素缺乏需使用生长激素治疗 [5] ,rhGH治疗可以显著提高GS患者的生长速度 [10] ,但基础疾病未得到诊治,患儿持续存在的电解质紊乱会继续降低生活质量。Junya Fujimura等人指出,GS患者的身材矮小和生长发育延迟与电解质紊乱有关,大多数身材矮小的患者在校正血清钾水平后,生长速度便可迅速改善,仍明显生长障碍的患者,需评估生长激素水平及青春期发育状态排除生长激素缺乏症 [12] 。因此鉴别矮小的病因,有利于正确评估病情、选择适当的治疗方案 [13] 。
低钾血症是最常见的电解质紊乱之一,病因繁多。了解低钾血症的常见病因,掌握其临床特点,可以减少漏诊、误诊。首先通过病史了解患者的家族史和伴随表现,以及是否存在钾摄入不足、胃肠道丢失或长期使用某些致血钾降低的药物;其次进行针对性的实验室检查,如电解质检测判断是否合并其它电解质紊乱,24小时尿电解质判断是否为肾性失钾;肾素、血管紧张素、醛固酮检测判断RAAS系统是否激活,血气分析鉴别肾小管酸中毒、GS、Bartter综合征(Bartter syndrome, BS)及Liddle综合征等。本例患儿表现有乏力、血压偏低,排除饮食、药物因素,无长期吐泻病史,存在低血钾、低血镁、低血氯、肾性失钾、高肾素,考虑GS或BS,完善基因检测进行鉴别,发现SLC12A3基因复合杂合突变,确诊为GS。遗传易感性是儿童低血钾的重要原因 [14] ,既往有研究指出,在无高血压的单基因低钾血症病例中,Gitelman综合征患者所占比例最大,分子诊断对指导精准治疗和改善疾病预后具有临床意义 [15] 。Sara S. Jdiaa等人指出,对于原因不明的低钾血症,详尽追问病史、密切关注临床及实验室证据对正确诊断疾病和制定适当的治疗方案至关重要 [16] 。
GS患者总体预后良好,但临床表现不典型,容易误诊、漏诊。该病例提示儿科医生,矮小病因复杂,诊治因疲劳、乏力、生长迟缓、身材矮小等问题就诊的患儿,应常规行血电解质检查,对持续低钾血症的孩子进行遗传学检查,做到对这类孩子早诊断、早治疗、改善长期预后,提高生活质量,促进优生优育。GS患者应个体化及终身替代治疗,长期规范管理和随访是薄弱环节,每年至少进行2次门诊随访,评估相关临床症状以及实验室指标,伴随生长迟缓的患儿要定期监测身高、体重,并根据病程和治疗效果调整治疗方案,达到最佳远期预后。
利益冲突
所有作者声明无利益冲突。
NOTES
*通讯作者。