1. 引言
近年来,全球乳腺癌发病率已居女性恶性肿瘤之首 [1] 。乳腺癌是性激素受体依赖的肿瘤之一,雌激素的存在可促进此类癌细胞的生长和增值 [2] [3] 。目前的方法尚不足以预测乳腺癌的治疗结果,即使是具有相同病历类型的患者,在经过手术与放射治疗后,预后也可能存在很大差别,这是因为乳腺癌具有异质性 [4] ,因此当前关于乳腺癌的分类标准还有待提高。而基因表达谱对肿瘤患者的分类和预后是有效的,在其海量数据中,使用数理统计模型有效地挖掘信息也受到业界越来越多的关注。对于分类问题,支持向量机、贝叶斯网络、人工神经网络、决策树等机器学习方法在各领域中都得到广泛的应用,并取得了良好的预测效果。但对于肿瘤患者来说,这些方法考虑了事件的结果而未充分应用出现这一结果所经历的时间,因此本文在此选择了对肿瘤患者更优的预后分组方法——Cox比例风险回归模型、LASSO以及生存树。本文利用乳腺癌基因芯片数据筛查影响乳腺癌患者预后的基因,通过筛选基因对患者进行分类可以发现不同类别患者的基因特征。在未来的乳腺癌治疗中,以期可对患者选择具有针对性的基因治疗方案,从而提高乳腺癌患者的生命质量。
2. 研究对象和方法
2.1. 研究对象
本文下载GEO数据库(https://www.ncbi.nlm.nih.gov/geo)乳腺癌基因芯片数据(GSE2034),该数据集共含有286个样本。其中209例为雌激素受体阳性(ER+)患者,该组患者随访时间为2个月至171个月,中位随访时间为86个月,本组80例患者出现复发;77例为雌激素受体阴性(ER−)患者,该组患者随访时间为6个月至161个月,中位随访时间为84个月,本组27例患者出现复发。上述患者均为淋巴结阴性,每个样本均含有22,283个探针。
2.2. 研究方法
本文分别对ER+乳腺癌样本和ER−乳腺癌样本进行研究。对于ER+乳腺癌患者,将209个样本随机分为训练集和测试集,其中训练集含90个样本,测试集含109个样本;对于ER−乳腺癌患者,将77个样本同样进行随机分组,其中训练集含42个样本,测试集含35个样本。训练集用于模型的建立,测试集用于检验训练好的模型的分辨能力。本文对ER+与ER−乳腺癌基因芯片数据的分析采用 R语言编程来实现。基因初步筛选使用单因素Cox比例风险回归模型,随后使用LASSO方法进一步筛选基因并建立生存树,使用Kaplan-meier曲线和对数秩检验对分类结果进行验证,以
为差异具有统计学意义。
3. 模型建立
3.1. 数据预处理
本文下载的乳腺癌基因表达矩阵如表1所示,行名表示探针,列名为每一例患者的编号;表2为患者基本信息。将两个表格通过患者编号进行合并,并通过“Status”将患者分为ER+乳腺癌组与ER−乳腺癌组分别进行研究。

Table 1. The microarray data of breast cancer
表1. 乳腺癌基因芯片数据
Table 2. The basic information of breast cancer
表2. 乳腺癌患者基本信息
将上述分组后的表格分别与表3所示的探针与基因匹配表结合进行整理,转换为每一个基因的表达值进行研究。若探针与基因为“一对一”的关系(即一个探针对应一个基因),则将相应表达值作为基因的表达值;若探针与基因为“一对空”或“一对多”,此时由于不能确定探针对应的是哪个基因的表达值,因此将其删除;若探针与基因为“多对一”,则取表达量较高的值作为此基因的表达值。经处理之后,ER+乳腺癌组与ER−乳腺癌组分别得到12,548和11,923个基因的表达值。
初始数据一般都具有冗余性、不完整性和不规范性,无法直接进行数据分析。一些无意义的数据的存在会严重影响算法的执行,若存在噪音干扰,还会造成结果的偏差。因此,对不理想的原始数据预处理是进行数据分析的首要步骤。为了去除芯片间的系统误差,本文对数据进行了分位数标准化 [5] ;同时为了减少背景噪音,将小于50的基因表达值赋值为50;接着对数据进行以2为底的对数化变换;再将变异系数小于3%的基因剔除,此时ER+乳腺癌组与ER−乳腺癌组分别剩余11,960和11,846个基因的表达值。最后对每一个基因表达值进行编码,计算每组全部基因表达值的25%、50%、75%分位数,小于等于25%分位数的编码为1,大于25%分位数且小于等于50%分位数的编码为2,大于50%分位数且小于等于75%分位数的编码为3,大于75%分位数的编码为4 [6] 。经过预处理的基因编码矩阵如表4与表5所示。
Table 3. The matching table of probes and genes
表3. 探针与基因匹配表
Table 4. The encoding matrix of ER+ breast cancer
表4. ER+乳腺癌基因编码值矩阵
Table 5. The encoding matrix of ER− breast cancer
表5. ER−乳腺癌基因编码值矩阵
3.2. 单因素Cox比例风险回归模型初步筛选基因
Cox比例风险回归模型 [7] 是由英国的生物统计学家Cox D R提出的比例风险模型。风险函数(hazard function)是描述生存时间分布的一个重要函数。如终点事件为死亡(复发),风险函数表示t时刻仍存活的病人在t时刻的瞬间死亡(复发)率:
称为瞬时死亡(复发)率或条件死亡(复发)速率。
Cox提出的比例风险模型是:病人具有
的伴随变量值,则第i名病人生存(复发)到时间t的风险函数是基础风险函数与自变量的函数的乘积:
即当所有的伴随变量都为0时的风险函数。其中定义伴随变量的函数
为指数形式,因此
Cox模型是一个风险比对数的线性模型,
实际意义为当伴随变量
每改变一个单位时所引起的相对风险度的自然对数的改变量。
不仅反映出协变量的作用强度,而且反映它的作用方向 [8] 。
我们对训练集数据使用单因素的Cox比例风险回归模型进行基因初步筛选,对于ER+乳腺癌组,以
作为入选标准。经过筛查,有999个基因对ER+乳腺癌的复发具有显著性影响,结果见表6 (结果保留两位小数)。如基因ABCB8,其b值为0.96,eb值为2.61,表明每当ABCB8的编码值增加一个单位,其复发风险比扩大到原来的2.61倍,说明对于ER+乳腺癌患者来说,ABCB8是其复发的风险基因;对于基因ABLIM1,其b值为−0.97,eb值为0.38,表明每当ABCB8的编码值增加一个单位,其复发的风险比缩小到原来的0.38,说明对于ER+乳腺癌患者来说,ABLIM1是其复发的保护基因。
对于ER−乳腺癌组,以
作为入选标准(在此分别使用
、
、
为限定条件对基因进行筛选,通过对最终结果进行比较,此步将P值严格限定为0.001)。经过筛查,有13个基因对ER−乳腺癌的复发具有显著性影响,结果见表7。
Table 6. Primary screening genes by univariate Cox proportional hazards regression model (ER+)
表6. 单因素Cox比例风险模型初步筛选基因(ER+)
Table 7. Primary screening genes by univariate Cox proportional hazards regression model (ER−)
表7. 单因素Cox比例风险模型初步筛选基因(ER−)
3.3. 计算风险分数,对患者进行分类
通过上一步的筛查,有999个基因与ER+乳腺癌患者复发显著相关,13个基因与ER−乳腺癌患者复发显著相关。本文使用所筛选的基因计算患者的风险分数,每例患者的风险分数为以相应Cox回归系数为权重的基因编码值的线性组合 [9] [10] 。接着使用风险分数对每一例患者进行高风险和低风险标记的划分,由于两组数据复发人数分别占每组人数的38.28%和35.06%,因此在此都选择风险分数的60%分位数作为对患者进行分类的阈值(≤60%分位数为低风险标记;>60%分位数为高风险标记),如表8与表9所示,其中0代表低风险标记,1代表高风险标记。
本文将训练集建模过程中得到的999个基因和13个基因的Cox回归系数和分类阈值直接应用于每组的测试集,计算测试集中每例患者的得分并进行高风险标记和低风险标记的划分。
最后,对分类结果进行检验,如图1与图2 Kaplan-meier曲线显示,两组数据的训练集和测试集中,低风险标记的患者相较于高风险标记的患者都有着较高的中位未复发时间。对数秩检验(log-rank test)显示,低风险标记与高风险标记的ER+乳腺癌患者的未复发率差异显著(训练集:
,
;测试集:
,
),低风险标记与高风险标记的ER−乳腺癌患者的未复发率差异同样也显著(训
Table 8. Calculate the risk scores and classify the ER+ breast cancer of training set
表8. ER+乳腺癌训练集患者风险分数及分类情况
Table 9. Calculate the risk scores and classify the ER− breast cancer of training set
表9. ER−乳腺癌训练集患者风险分数及分类情况
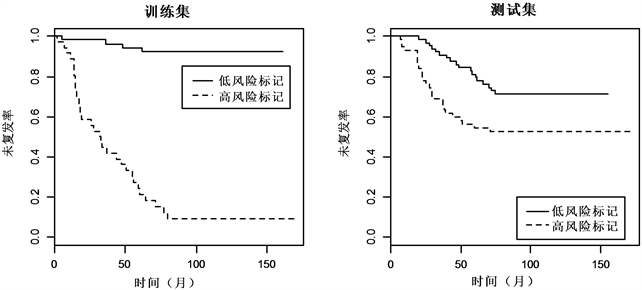
Figure 1. Kaplan-Meier estimates of survival of ER+ breast cancer according to the 999-gene signatures
图1. ER+乳腺癌999-基因标记Kaplan-Meier曲线
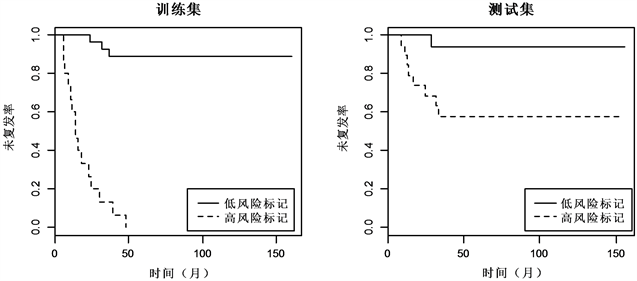
Figure 2. Kaplan-Meier estimates of survival of ER− breast cancer according to the 13-gene signatures
图2. ER−乳腺癌13-基因标记Kaplan-Meier曲线
练集:
,
;测试集:
,
)。
3.4. LASSO方法筛选基因
由于数据中基因数量多而样本量较少,并且各基因间可能存在交互作用,因此选用LASSO方法对基因进行进一步筛选。LASSO对于高维度、强相关、小样本的生存资料数据较为适用。LASSO的基本思想是在回归系数的绝对值之和小于一个常数的约束条件下,使残差平方和最小化,从而使某些回归系数严格等于0,来得到可以解释的模型 [11] [12] 。
该方法的估计参数
随着l的增加,
项就会减小,这时候一些自变量的系数就逐渐被压缩为0,以此达到对高维资
料进行降维的目的。LASSO方法的降维是通过惩罚回归系数的数量来实现的。
交叉验证法 [13] 是比较常用的推测估计调整参数l的方法,如k折交叉验证。在k折交叉验证中,所有的数据观测值被大致分为k等份,然后轮流以其中所有可能的k-1份为训练集对数据进行拟合,剩下1份作为测试集,即测试集与训练集的观测值数目之比约为1:(k-1),一共计算k次,得到拟合测试集时的误差率(或其他指标)那样的k个指标,再做平均。在对每个模型都做一遍之后,最后选择误差率最小的模型。
我们选择10折交叉验证的方法来估计调整参数l。对于ER+乳腺癌组,当
时,误差率取得最小值,此时进一步筛选出了5个基因:CCDC69、KIF18A、KIF23、PLA2G15、RAI2;对于ER−乳腺癌组,当
时,误差率取得最小值,此时进一步筛选出了10个基因:ABCC2、AKT1、ATP4B、CD200、FICD、PARP4、POLDIP2、RECQL5、THPO、XRCC1。
3.5. 生存树方法进行预测
生存树方法是由Gordon和Olshen [14] 在分类与回归树的基础上改进而成的,不同于普通的分类树,其对截尾生存资料同样适用。通过递归分割计算,树逐渐进行生长。即选择某一截断点将根节点分为两部分,在分开的两个子样本中,其生存分布差异达到最大,即两组人群的预后相差达到最大。最优划分
满足
这里,
表示两样本log-rank检验统计量,其中s表示节点h内所有可能的截断方式,重复应用这个规则进行划分。
树的停止规则为:1) 结点内样本例数太小;2) 划分函数的测度不够充分,即划分得到的两个子结点的生存分布无差别。这时就得到了一棵初始树(initialtree) [15] 。一般情况下初始树过大,会产生过度拟合的现象,在对未来样本进行预测时较为不准确,而且不容易解释,因此,需要通过剪枝过程对初始树的节点进行删减,控制树的复杂度 [16] 。
对于ER+乳腺癌,本文使用所筛选的5个基因对训练集数据建立生存树模型,并将结果应用于测试集,得到了如图3所示的一棵生存树。将第1、2、3、4、5、7个叶节点作为低风险标记,第6、8、9个叶节点作为高风险标记。
对于ER−乳腺癌,本文使用所筛选的10个基因对训练集数据建立生存树模型,进一步筛选出了5个基因:AKT1、CD200、FICD、THPO、XRCC1。将此结果应用于测试集,得到了如图4所示的一棵生存树。然后将第1、2、3、5、6个叶节点作为低风险标记,第4、7、8个叶节点作为高风险标记。
同样,对分类结果进行检验,如图5与图6 Kaplan-meier曲线显示,两组数据的训练集和测试集中,低风险标记的患者相较于高风险标记的患者都有着较高的中位未复发时间。对数秩检验显示,低风险标记与高风险标记的ER+乳腺癌患者的未复发率差异显著(训练集:
,
;测试集:
,
),低风险标记与高风险标记的ER−乳腺癌患者的未复发率差异同样也显著(训练集:
,
;测试集:
,
)。
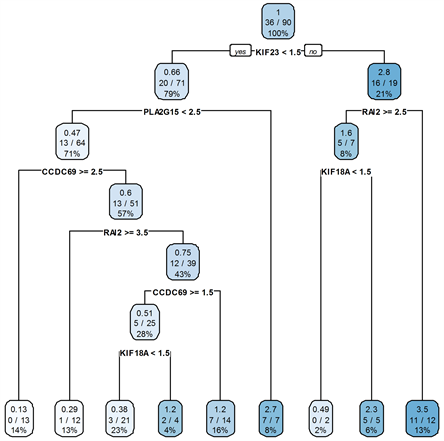
Figure 3. Survival tree of ER+ breast cancer
图3. ER+乳腺癌生存树
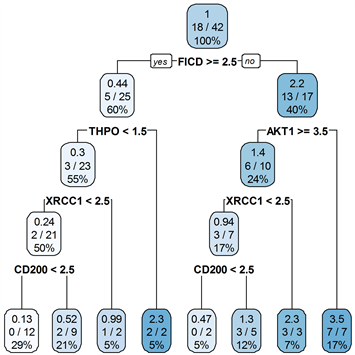
Figure 4. Survival tree of ER− breast cancer
图4. ER−乳腺癌生存树
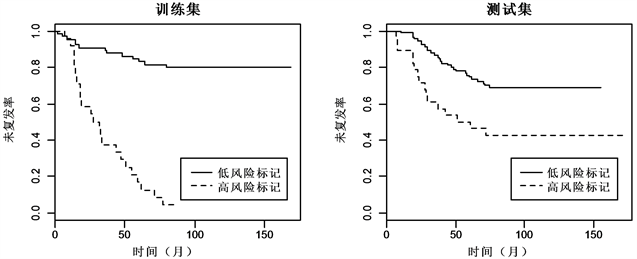
Figure 5. Kaplan-Meier estimates of survival of ER+ breast cancer according to the 5-gene signatures
图5. ER+乳腺癌5-基因标记Kaplan-Meier曲线
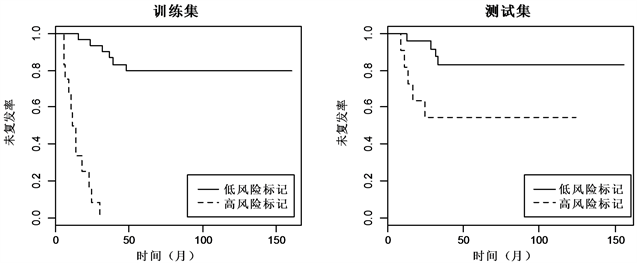
Figure 6. Kaplan-Meier estimates of survival of ER− breast cancer according to the 5-gene signatures
图6. ER−乳腺癌5-基因标记Kaplan-Meier曲线
4. 讨论
本研究对互联网上下载得到的乳腺癌基因芯片实验数据建模分析,分别对ER+乳腺癌和ER−乳腺癌患者的复发进行了研究,筛选对其复发具有影响的基因。对于高维生存数据,采用Cox比例风险回归模型进行了初步筛选,经检验,由此步所筛选基因编码值的线性组合所进行的分类效果差异显著。由于我们希望可用较少的基因达到较优的预测效果,因此继续进行下一步筛选。LASSO方法在处理高维数据的生存分析方面效果显著,因此将由Cox比例风险回归模型筛选出的基因应用到LASSO方法分析,得到了对ER+乳腺癌和ER−乳腺癌复发有显著性影响的基因,且各基因间可能存在交互作用。我们将其应用于生存树对患者进行高风险标记与低风险标记的分类。为了验证所建立模型的有效性,本文使用测试集数据进行了检验,最终在对ER+乳腺癌与ER−乳腺癌两组数据复发风险的预测中,结果均具有统计学意义,且得到如下结论:
1. CCDC69、KIF18A、KIF23、PLA2G15、RAI2对ER+乳腺癌的复发具有显著性影响;
2. AKT1、CD200、FICD、THPO、XRCC1对ER−乳腺癌的复发具有显著性影响。
在ER+乳腺癌最后筛选的5个基因中,有2个基因在相关文献中已被证明在乳腺癌中发挥着重要作用。Zhang C,Zhu C等 [17] 的结果表明Kif18a参与乳腺癌中且可作为乳腺癌的潜在治疗靶点;Werner S,Borgmann K等 [18] 发现RAI2功能的丧失与有丝分裂保真度降低有关,除了维持激素依赖性乳腺肿瘤的分化外,RAI2还作为维持基因组完整性的一般肿瘤抑制因子。在ER−乳腺癌最后筛选的5个基因中,有3个基因在相关文献中已被证明在乳腺癌中发挥着重要作用。Kabraji S, Solé X等 [19] 表明AKT1低沉降癌细胞可能成为三重阴性乳腺癌患者新辅助化疗后持续存在的非遗传细胞状态,并值得进一步研究。Spears M,Cunningham C A [20] 的研究也显示,AKT1激活与早期乳腺癌的不良预后有关。Erin N,Podnos A等 [21] 的研究结果支持CD200表达可以改变免疫反应的假说,并且可以抑制诱导全身和局部炎症反应的肿瘤细胞的转移生长。增加CD200活性/信号可能是治疗侵袭性乳腺癌的重要治疗策略。Moullan N,Cox D G等 [22] 的研究显示XRCC1基因多态性的不同的组合似乎与乳腺癌风险增加有关,或与某些乳腺癌患者出现不良放疗反应的可能性相关。
CCDC69、KIF23、PLA2G15这3个基因在ER+乳腺癌中发挥的作用及FICD和THPO在ER−乳腺癌中发挥的作用仍需通过进一步生物实验验证,其可能为ER+乳腺癌和ER−乳腺癌复发的潜在影响因子。总之,由我们筛选的基因对ER+与ER−乳腺癌患者风险高低的分类与患者临床结果密切相关。希望这些基因能为进一步实验提供理论依据,以研究这些基因在乳腺癌的预后中发挥的潜在作用。
基因项目
国家自然科学基金(11271125)。