1. 引言
蛋白质复合物是由一组具有功能相似的蛋白质组成,它是细胞组织形成和功能实现的关键细胞实体。因此,预测蛋白质复合物可以从系统水平上更好地理解细胞的基本组成和组织机制。通过实验方法检测蛋白质复合物通常比较昂贵并耗费大量时间。而且,有些复合物只能在特定的条件下才能被检测。近年来,高通量实验技术和机器学习的方法产生了大量的蛋白质相互作用数据(PPI)。基于复杂网络的聚类技术、机器学习理论和群智能算法 [1] 的应用为蛋白质复合物识别奠定了理论基础和技术指导。因此,设计有效的聚类方法识别相互作用网络中的复合物是生物网络分析的一个关键问题。
复合物主要特征:1) 复合物内蛋白质具有结构和功能一致性;2) 在拓扑方面复合物具有相对较高的密度。复合物识别任务转化为利用聚类算法从图中识别蛋白质节点集合。整个PPI网络可以转化为一个图 [2] 。根据网络的性质,将其分为有权网络和无权网络,在有权PPI网络中,两个节点之间边的权值由这两个节点之间存在的相互作用关系的可能性大小表示。在无权的PPI网络中,所有边权值相同,一般使用二进制的邻接矩阵表示蛋白质节点之间的关系,如果两个蛋白质节点之间没有相互作用关系则用0表示,否则用1表示。研究发现,在有权网络上进行实验更容易识别出蛋白质复合物。
近年来,复合物识别算法通常基于图的算法聚类PPI网络。这些方法主要基于PPI中拓扑属性识别复合物,包括:MCODE [3] 、CFinder [4] 、MCL [5] 、FLCD [6] 和CDRWR [7] 。MCODE将PPI网络中具有相对高密度的区域定位为蛋白质复合物。CFinder基于团渗透方法在PPI网络中识别重叠蛋白质复合物。文献 [5] 引入马尔可夫聚类算法识别PPI网络中高度联通的蛋白质复合物。文献 [8] 引入随机游走马尔科夫聚类实现在有权或无权图图中识别蛋白质复合物。FLCD将PPI网络转化为有向无环图,确定子图的搜索空间,能够在加权/无权PPI网络挖掘网络中高连通子图。CDRWR使用相似性计算PPI中边的权值,并扩展重要种子节点以确保复合物的完整性,然后通过密度选择外部节点,通过合并策略形成最终的复合物。这些方法通常只关注单一的网络拓扑特征,其使用的PPI数据中含有的噪声影响预测结果。基于这些不足和复合物的特征,本文提出一种基因本体和核-附属结构的蛋白质复合物识别算法,主要工作包括:1) 通过语义相似性融合蛋白质相互作用网络和基因本体信息构建有权图模型;2) 设计以局部子图直径和密度为聚类条件的核-附属结构的复合物识别算法GCA;3) 比较并分析GCA与三个经典的方法,实验结果表明,GCA在召回率、f度量和功能富集分析方面的表现均显著优于CFinder,MCode和MCL。
2. 基于基因本体和核-附属的蛋白质复合物识别算法
本文中PPI网络用无向有权图
表示,其中V是网络中蛋白质构成的节点集合,E是相互作用的蛋白质构成的有权边集合。蛋白质复合物识别工作包括有权网络模型构建和基于核-附属的复合物识别算法。
2.1. 有权网络模型构建
有效的考虑网络权重可以进一步提升蛋白质复合物识别的性能。基因本体(GO)是跨物种的综合资源,描述与生物过程,分子功能和细胞成分相关的基因和基因产物生物学特性,提供了蛋白质对之间的功能关系 [9] 。本文首先使用语义相似性构建基于基因本体和蛋白质相互作用信息的有权网络模型,通过最佳匹配(BMA)策略 [10] 计算每个行和列的所有最大相似度的平均值 [11] ,用于计算图中边的权重,如公式(1)-(3),其中A/B分别表示DIP数据中蛋白质对应的基因,
表示由GO注释得到关于蛋白质A的集合,
表示由GO注释得到关于蛋白质B的集合。
(1)
(2)
(3)
其次,基于公式(4)~(6),根据基因本体的分子功能、生物过程和细胞组成三个不同特性,分别构建基于分子功能的有权图模型M,基于生物过程的有权图模型O,基于细胞组成的有权图模型C。最后,根据公式(7),采用均值方式融合三个模型M,O和C,构建基于基因本体的有权网络模型G。
(4)
(5)
(6)
(7)
2.2. 基于核-附属的蛋白质复合物识别模型
蛋白质网络中相同簇中的蛋白质通常比外部簇中的蛋白质具有更紧密地相互作用;生物网络具有小世界的特性。因此,在复合物识别的聚类过程中,采用子图密度和子图直径为聚类约束条件,其中密度计算如公式(8),
是有权图中节点v的度,
是网络中的边e的权值,
为有权网络中节点的个数。
(8)
算法过程描述如下

算法包括四个步骤:节点度的计算和排序,种子节点的选取,核心簇的形成以及核心-附属复合物的形成。其中核心簇的形成由两个约束条件确定:密度和直径。如果以当前节点为种子节点形成簇的过程中满足条件(9),则将其加入到当前簇中;否则继续找其他节点,直到不满足条件(9)为止,形成一个核心簇。然后选择新的种子节点,重复这个过程,直到形成所有的核心复合物。根据文献经验得知 [12] [13] ,通常设置
,
。核心-附属算法以蛋白质复合物通常具有高度相互作用的蛋白质的密集核心理论的基础。此类模型识别步骤分两步,首先发现网络中高度连接区域,然后通过添加强关联邻居来扩展这些区域。
(9)
3. 实验与结果分析
3.1. 数据集及评价标准
文中实验使用CYC2008 [14] 和MIPS [15] 作为基准复合物数据集评估不同方法预测的复合物的性能。酵母蛋白质相互作用数据DIP [16] 和基因本体论(GO)信息用于构建有权图模型。
给定一组已知的真实蛋白质复合物集合
和一组已识别到的蛋白质簇集合
,如果定位到的蛋白质簇
与真实蛋白质复合物
满足条件
,则认为
和
是匹配的。
是识别的蛋白质复合物
和真实蛋白质复合物
之间交集的大小,
和
分别为检测到的复合物集合I的大小和真实的复合物集合R的的大小。为了评估预测算法的性能,采用recall,precision和f-measure三个评测指标,如(11)~(13)。
(10)
(11)
(12)
(13)
3.2. 实验结果分析
为综合比较算法GCA的复合物识别性能,本文将GCA与CFinder,MCode和MCL三个算法就各自算法的准确率,召回率和f度量进行了比较。3种方法的参数都采用文献中提供的默认参数值。如图1在数据集CYC2008上的表现性能所示,在准确率方面,GCA弱于MCode,说明本算法在噪声数据去除方面还有进一步的提升空间。但是综合蛋白质复合物识别性能评测的3个指标,GCA在蛋白质识别的召回率和综合指标f度量方面都是最优的。召回率比较说明,本文算法GCA识别的复合物能够匹配的真实的复合物的数量最多。在f度量方面分别高于CFinder,MCode和MCL的百分点为12%,21%和17%。这说明与图中其他的复合物识别算法相比,本文算法在匹配真实复合物数量,综合表现性能方面具有更好的优势。分析图2可以得出类似结果。
3.3. 不同阈值t条件下算法性能比较
为了进一步说明GCA算法的有效性,图3和图4列出了四种算法在9个不同阈
条件下f度量的对比结果。从图3可以看出,本文提出的蛋白质复
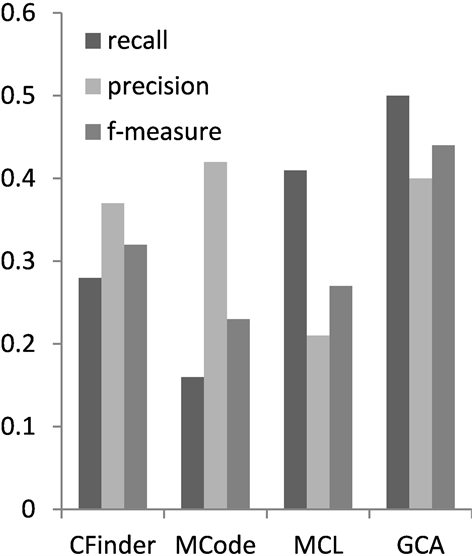
Figure 1. Performance comparison on CYC2008
图1. 在CYC2008上性能比较
合物识别算法的匹配性能要显著优于其他算法。当匹配的阈值越低,算法复合物识别的f度量综合性能越高。当阈值越高,算法在复合物网络中f度量表现性能逐步下降。特别是在数据集MIPS上GCA较其
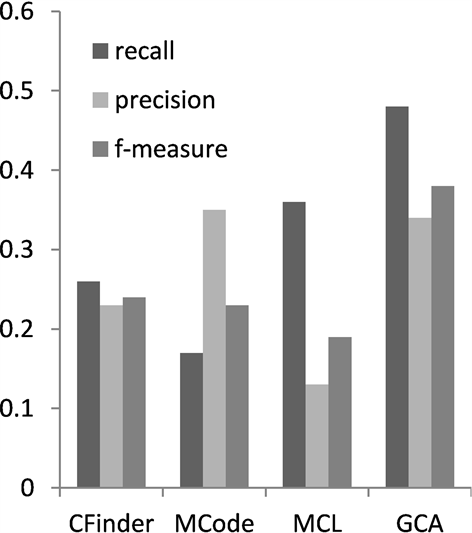
Figure 2. Performance comparison on MIPS
图2. 在MIPS上性能比较
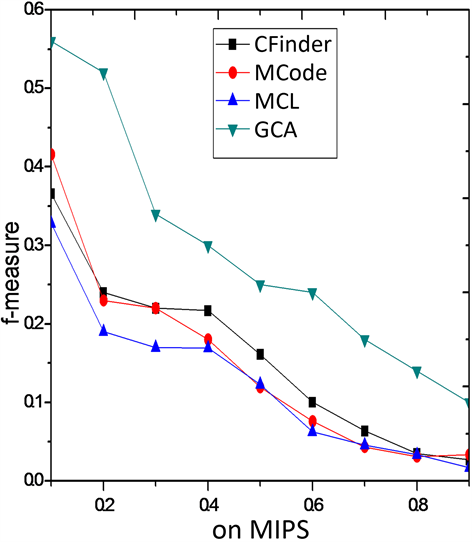
Figure 3. F-measure comparison on MIPS
图3. 在MIPS上F-measure性能比较
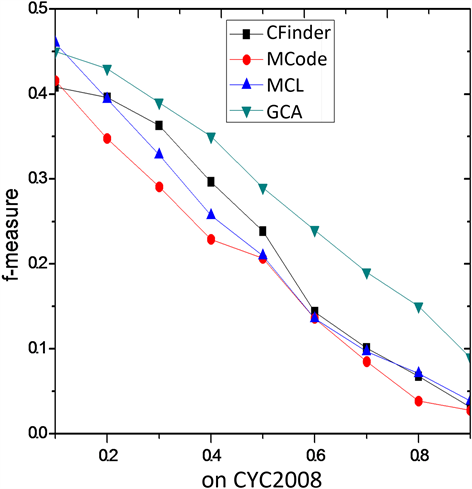
Figure 4. F-measure comparison on CYC2008
图4. 在CYC2008上F-measure性能比较
他算法优势更加明显。图4在CYC2008上的比较结果与图3类似。综合分析,GCA算法在酵母菌的网络DIP上的f度量表现整体性能要优于其他三类算法,具有较强的蛋白质复合物识别的能力,进一步说明引入基因本体的核-附属算法的有效性。
3.4. 功能富集分析
实验通过计算每个识别出蛋白质复合物的
值可以分析识别到的复合物所具备的生物意义。
值反映的是蛋白质复合物所包含的某一特定生物功能的富集程度,可以注释出该复合物所具备的主要功能。
值的计算公式如(14):
(14)
其中,
表示识别的蛋白质复合物中蛋白质的个数,l表示复合物C中具有某一功能x的蛋白质个数。
表示蛋白质网络中蛋白质的个数,其中具有功能x的蛋白质的个数为
。
体现了识别复合物中蛋白质功能富集的概率。如果复合物被注释成好几个功能,则只取
最小时对应的功能。
本实验对从酵母PPI网络中识别蛋白质复合物进行功能富集分析进一步验证GCA算法的有效性,具体
值的分析和比较如表1。每个复合物的
的范围从小到大划分成四个区间:< E-15,[E-15, E-10],[E-10, E-5],[E-5, 0.001]。
值大于0.001,通常认为复合物的功能被随机指派的可能性很大,基本没有生物意义。表1中括号内的百分数表示
落在某个区间的复合物数与落在所有区间的复合物数的比值。例如GCA总共预测到219个复合物,其中
落在区间 < E-15的复合物数占百分比为21.75%。从表1中可以发现,在
最小的区间(即 < E-15),GCA预测的复合物无论数量还是百分比都远远大于其他算法,如前所述,
越小表示复合物的生物意义越显著,因此,这些复合物都具有最显著的生物意

Table 1. Comparison of functional enrichment
表1. 功能富集性比较
义。GO功能富集分析都说明GCA算法性能优于比其他算法。
4. 结论
在PPI网络中检测蛋白质复合物是生物医学领域的重要任务。因此,随着技术的进步,PPI网络的增长速度比以往任何时候都快,这使得任务变得非常重要。在本文中,我们提出了一种基于基因本体和核-附属的有权图聚类方法,通过融合基因本体和蛋白质相互作用数据构建有权图模型,设计基于核-附属的算法进行复合物识别,可以有效地从复杂的网络中找出密集连接的子图。与CFinder,MCode和MCL相比,本文中提出的算法可以挖掘更多的蛋白质复合物,提高算法的识别性能。在今后进一步的研究工作中,我们将考虑有差异的利用不用的数据源信息构建有权图模型,并设计基于分类型的聚类识别方法。
基金项目
辽宁省教育厅科技项目(L201605)。