1. 引言
蛹虫草又名北冬虫夏草、北虫草,与冬虫夏草同属异种,是虫草属的模式种 [1],经成分测定分析,在药化、药理和临床实验方面被证明完全可以作为冬虫夏草的代用品 [2]。蛹虫草中主要活性成分如虫草素、虫草酸及虫草多糖等,具有药用价值和保健功能。近年来,蛹虫草开始受到关注,并作为非处方药使用。2005年4月,蛹虫草被正式收录于《中华人民共和国药典》,2009年3月被正式批准为新资源食品 [3],2014年10月改称为新食品原料 [4]。蛹虫草是目前公认的食药用虫草之一,在我国已经形成了一个巨大的产业,年产值可达100亿元人民币 [5]。蛹虫草菌分有性(异宗配合) [2] 和无性(蛹草拟青霉) [6] 两个生育阶段。人工蛹虫草多采用无性繁殖,蛹虫草菌有若干菌株,不同菌株生产的虫草,质量高下差别大 [7]。从事蛹虫草人工栽培研究工作的科研单位及生产基地,都在不断探索和研究在大规模生产栽培中提高产量和活性成分含量的有效途径。基因组学为解决蛹虫草各种生物活性物质的功能基因和其产业化奠定了基础,随着基因组学的深入研究和当今生物信息学的飞速发展,蛹虫草神秘的面纱将一步步被揭开 [8]。
测序技术飞速发展极大地促进了基因组结构和功能元件的研究,分子生物学方法和技术在解决物种的亲缘关系、起源与进化、遗传与变异等问题中起了重要作用 [9],2011年,蛹虫草基因组得以破译 [5],组装后基因组大小为32.2 Mb,GC含量为51.4%,含7条染色体,大小在2.0~5.7 Mb之间;基因组中共含有9684个蛋白编码基因,超过63%的编码基因在菌丝体和子实体发育阶段表达,大约16%的编码基因(1547个)参与菌物–昆虫的相互作用,不存在编码对人类有害的已知真菌毒素的基因 [9],蛋白质编码基因中13.7%为种特异性的基因。然而,这些基于DNA序列的基因组数据并不能反映基因组上编码区和非编码区的转录水平和转录调控机制 [8]。因此,从RNA水平研究真核生物在特定生长时期和培养条件下全基因组转录情况的转录组学为这一问题提供了解决方案 [10]。它通过研究特定环境条件下基因编码RNA (mRNA)和非编码RNA的表达水平及其调控规律,进一步推动了基因功能和ncRNA调控机制的研究 [11]。
2. 测序流程及生物信息学分析策略
不同微生物基因组GC含量差别很大,这种差别对基因及相应蛋白质的组分具有较大影响。另一方面,特定蛋白质为了保持其结构和功能,对蛋白质编码序列在单核甘酸、密码子、氨基酸和具有相同理化性质的氨基酸等不同层次上具有限制作用 [12]。分析基因组GC含量对蛋白质组分的影响对理解自然选择与碱基突变的关系以及微生物进化具有重要意义。
2.1. 测序策略
Solexa高通量测序技术是以单分子阵列技术为基础,是对合成测序技术的发展与延伸。Solexa是一种基于边合成边测序技术(Sequencing-by-Synthesis, SBS)的新型测序技术。通过单分子阵列实现在小型芯片(Flow Cell)上进行桥式PCR反应。通过可逆阻断技术实现每次只合成一个碱基,再利用相应的激光激发荧光集团,捕获激发光,从而读取碱基信息 [13]。Illumina HiSeq测序,提供不低于基因组100×的Illumina测序数据量,组装获得扫描图序列,并进行后续分析 [14]。
2.2. 生物信息学分析策略
测序完成后对序列进行数据统计分析及生物信息学分析 [15],测序工作得到数据仅仅是第一步,进一步的分析和挖掘这些信息中所蕴含的生物学意义才是最终的目的。比较基因组学就是通过比较序列间碱基或氨基酸的差异,获取有关进化的深层次的信息,进而构建进化树研究它们之间的同源性 [16]。近年来,较多的模式生物基因组测序任务的完成,为从整个基因组的角度研究分子进化提供了有力的条件,分析策略见图1。
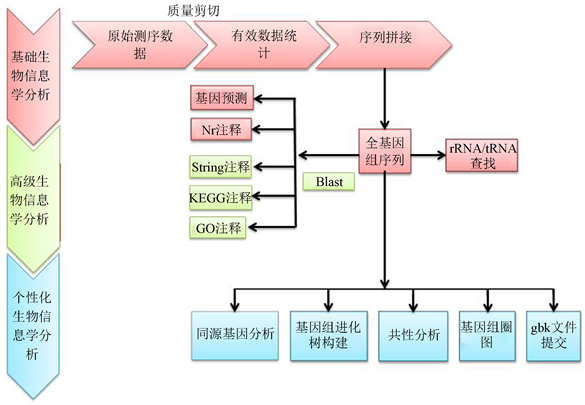
Figure 1. The bioinformatics analysis strategy of genome
图1. 基因组生物信息学分析策略
3. 生物信息学分析解析
3.1. 基因组评估
对初步组装得到的基因组序列进行质量评估,目的如下:① 判断基因组是否存在污染。一些杂菌的污染在进行测序前质检时可能无法检测出来,比如一些亲缘关系较近的杂菌的污染,或占比较低的轻度污染等,会对基因组的精确组装和后续分析造成干扰,因此需进行评估,以保证结果的准确性。② 评估测序质量③ 评估基因组情况。
通过GC_depth分布图可以看出测序是否有明显的GC偏向,也可以判断是否存在污染等情况。一般情况下,相对于中等GC含量区域来说,高GC含量区域或者低GC含量区域的测序深度都比较低。通过GC_depth分布图可以看出测序是否有明显的GC偏向,也可以判断是否存在污染等情况 [17]。一般情况下,相对于中等GC含量区域来说,高GC含量区域或者低GC含量区域的测序深度都比较低;利用PE测序reads,选取中间高质量测序区域,逐碱基取某一长度为K-mer进行基因组大小评估。以统计各17-mer深度和各个深度的频数所占比例为例,计算作图获得深度–频率分布图 [18]。该分析可对真核类基因组进行杂合度、基因组大小评估。
3.2. 基因组组装与预测
利用质控后的符合要求的clean data进行从头组装得到基因组序(scaffold),对组装序列进行基因预测 [19],得到每个样本的基因信息。
3.2.1. 基因组组装
利用短序列组装软件对优化序列进行多个K-mer参数的拼接,得到最优的组装结果,相关结果参数见表1。

Table 1. Results parameters for genome assembly
表1. 基因组组装的结果参数
注释:N50、N90长度的概念:将各个序列按长度大小排序,从大至小逐一扫描各个序列的长度值,进行累加,当该累加值第一次超过所有序列总长的50%时,此时扫描到的序列,其长度值即为N50值,N90值亦同理。N50、N90长度值比平均长度更能准确表示拼接序列的好坏。Contig:重叠群,拼接软件基于reads之间的overlap区而拼在一起的序列,中间没有Gap。Scaffold:框架序列,基于paired-end文库的序列信息,确定contigs之间的顺序关系,将contigs按顺序排列在一起形成的更长序列,即为scaffold,中间可能有Gap。
3.2.2. 编码基因预测
利用maker2软件进行真菌的基因预测,对基因组中的编码序列(CDS)进行预测结果参数见表2,获得功能基因的核酸序列和氨基酸序列 [20] [21],用于后续功能和系统进化分析。
3.2.3. 基因注释
对预测得到的编码基因进行基础的功能注释,通过与5大数据库(NR库,swiss-prot库,Pfam库,COG数据库,GO数据库)进行比对进行功能注释 [22]。基因注释主要是基于蛋白序列比对。将基因序列与各个数据库进行比对,得到对应的功能注释信息。
Table 2. Result parameters of coding gene prediction
表2. 编码基因预测的结果参数
(1) NR/Swiss-prot数据库注释分析:NR数据库是NCBI推出的整合型蛋白数据库,所包含的蛋白序列信息相对丰富。而Swiss-prot数据库是Uniprot Knowledge-Base推出的高质量数据库 [23],虽然其所含序列信息少于NR数据库,但其包含的是经过人工认证过的序列,可信度比较高。根据实验样本特性和分析目的,可以灵活选用数据库来达到更好的分析效果。
(2) COG功能分析:COG是Clusters of Orthologous Groups of Proteins的缩写。进行COG数据库比对可以对预测蛋白进行功能注释、归类以及蛋白进化分析。
(3) KEGG功能分析:在生物体内,基因产物并不是孤立存在而各自发挥作用的,不同基因产物之间通过有序的相互协调来一起行使具体的生物学功能。因此,KEGG数据库中丰富的通路信息将有助于我们从系统水平去了解基因的生物学功能 [24],例如代谢途径、遗传信息传递以及细胞学过程等一些复杂的生物过程。
3.2.4. 结构基因组查询
基因结构查询从基因组整体层面进行分析,包括启动子预测,基因组图谱绘制,通过结构分析可以更全面的掌握该样本基因组情况。
(1) 启动子预测:启动子是调控基因表达的调控元件,决定了基因表达的强度和时机,通过启动子的插入或缺失,可以改变基因的表达模式,实现对菌体生长发育以及代谢的调控研究 [25]。启动子预测结果以基因为单位,展示相应基因的启动子序列,包括启动子的位置、长度、序列信息等,此外可以通过筛选功能直接获得某个基因或基因组某个区域内的启动子信息,结果参数见表3。
(2) 基因组图谱:真菌组装结果是scaffolds水平,通过线性图展示每条scaffold上编码基因组的排列情况,标注每个基因的编码方向,用颜色代表每个基因的COG功能分类。通过筛选功能可以搜索某个基因名称,展示相应scaffold的图谱,从而查看目的基因上下游的基因,对相关的研究分析具有重要意义。
(3) 基因功能查询:嵌入各种基因查找小工具,通过基因名称或关键字搜索就可以获得相应基因的详细信息,综合基因多个注释库信息,对基因进行多维度数据挖掘。
3.2.5. 代谢系统分析
(1) 碳水化合物活性酶注释:碳水化合物活性酶数据库是关于合成或分解复杂碳水化合物和糖复合物酶类的专业数据库,根据蛋白质结构域中氨基酸序列的相似性,可将不同物种来源的碳水化合物活性酶分成糖苷水解酶,糖基转移酶,多糖裂合酶,碳水化合物酯酶,碳水化合物结合模块,辅助氧化还原酶等六大类蛋白质家族。
Table 3. Result parameters of promoter prediction
表3. 启动子预测的结果参数
(2) 细胞色素P450注释:细胞色素P450 (Cytochromes P450,简称CYP450)是一大类以亚铁血红素为辅基的蛋白家族。它们能催化许多种底物的氧化反应。因这类蛋白的还原态与CO结合后会在450 nm处检测到最大吸收波长,故又命名为P450。它参与内源性物质和包括药物、环境化合物在内的外源性物质的代谢。
3.2.6. 比较基因组分析
(1) 同源基因分析:采用OrthoMC软件对不同蛹虫草菌株的氨基酸(或核苷酸)序列进行比对,选取一定阈值(阈值一般在30%~80%之间,视具体项目情况而定)进行相似性聚类,获得同源基因的列表 [26]。统计每一个蛋白聚类cluster的物种分布情况,可以进行属内或种内的泛基因组、核心基因组的研究。
(2) 系统发育树分析:基于同源基因分析的结果,选取单一拷贝的同源基因进行多序列比对和比对质量控制。然后基于单拷贝基因法构建系统发育树(图2)。
(3) 基因簇比较分析:微生物基因簇是一组紧密连锁的且功能上密切相关的结构基因,这些基因各自编码的酶常能组成多酶复合物,能催化代谢中的特定反应。基于已知的基因簇信息,通过基因簇比较作图分析,可以直观的了解研究的菌株间的基因簇信息。
(4) 共线性分析:共线性一般是指不同物种中染色体之间的某些区域有着相似的基因排列顺序。一般而言,进化距离越远的物种之间基因共线性越差 [27],因此两个物种之间的共线性程度可以作为衡量它们之间进化距离的标尺 [28]。同时,我们还可以通过共线性分析来获得物种间基因组在进化过程中所发生结构变异的情况。
(5) 泛基因组分析:泛基因组是指某一类物种(或菌株)全部基因总和,包括核心基因组和非必需基因组 [29]。核心基因组是在该类物种(或菌株)中都存在的基因,一般与必需的生物学功能和重要表型特征相关,反应物种的稳定性。非必需基因组是指某一(或以上)种物种(或菌株)中存在的序列,一般与蛹虫草菌对特定环境的适应性或特有的生物学特征相关,比如产虫草素、耐受性和退化等,反映了这类物种(菌株)的表型特性 [30]。
4. 问题与展望
蛹虫草与其它虫草不同之处在于其具有较高含量的虫草素,虫草素又称虫草菌素,化学名为3’-脱氧腺苷,是第一个从真菌中分离出来的核苷类抗生素 [31]。由于其具有杀虫、抗菌、抗病毒、抗肿瘤、抗氧化、抗衰老、免疫调节等广谱的生物学活性 [32],近70年来在药物化学领域的研究一直非常活跃,不同药理功能的分子作用机理正在逐步获得清晰的解析,并且在应用研究方面都取得了开创性的成果。虫草素多样的药理作用已经得到了公认,无论是在医药还是保健食品方面,都拥有巨大的开发价值和广阔的市场前景 [33]。随着基因组的破译,蛹虫草分子方面的神秘面纱在逐步揭开,一系列关于蛹虫草转录组学、蛋白质组学等组学方面的研究也取得了较大进展。然而,还有许多问题没有得到解决 [34]。虫草素作为蛹虫草中最重要的一类活性物质,目前市场上的虫草素主要是通过人工培养的蛹虫草子实体获得,但蛹虫草属营养缺陷型真菌 [35],各蛹虫草菌株的虫草素产能参差不齐,普遍较低,且遗传稳定性较差,导致其市场价格昂贵,己经成为制约蛹虫草规模化稳产和虫草素开发利用的瓶颈 [36]。随着基因组学的深入研究和当今生物信息学的飞速发展,改良选育优质高产的虫草素生产菌株,定向开发高产虫草素的蛹虫草无疑是今后的主要发展方向。
基金项目
江西科技师范大学大学生创新训练计划项目(20191304160)。
NOTES
*通讯作者。