1. 引言
为了缓解环境污染、能源短缺等问题,清洁能源的探究使用和转换储存技术有待进一步研究 [1] [2]。燃料电池可以将化学能直接转换成电能储存备用,是一种高效、环境友好的能源转化器件 [3] [4]。但是,其阴极的氧气还原反应(oxygen reduction reaction, ORR)动力学进程缓慢,极大地限制了燃料电池的整体效率。因此,制备具有高氧气还原活性和高稳定性的阴极ORR催化剂至关重要 [5]。目前,碳基材料因其良好的导电性、大的比表面积、廉价易得等优点引起广泛研究 [6] [7]。石墨相氮化碳具有类似于石墨的层状稳定结构,成本低、操作简单,但由于其属于半导体材料、导电率较差,限制了其在电催化氧气还原方面的实际应用 [8] [9] [10]。为了解决石墨相氮化碳存在的问题,目前主要的解决方法是将石墨相氮化碳负载在导电基底上以提高其氧气还原性能 [11] [12] [13] [14]。Yang等人 [15] 制备了石墨烯基氮化碳纳米片,该复合材料表现出高氮含量、高表面积和增强的导电性。使其作为ORR无金属催化剂时,展现出优异的电催化活性、良好的耐久性和高选择性。Sarkar等人 [16] 合成的Cu-g-C3N4材料在碱性介质中对ORR表现出高甲醇耐受性、长期稳定性。Park等人 [17] 制备了石墨氮化碳–碳纳米纤维(g-C3N4-CNF),该催化剂对氧气还原反应和氧气析出反应显示出高的催化活性,可作为良好的双功能催化剂。上述学者提出的方法提高了g-C3N4的氧气还原性能,但是仍然存在一些问题:在严苛环境下活性容易退化和遭受腐蚀、碳纳米纤维等材料的制备成本较高等。因此,制备一种导电性能好、化学性质稳定、成本低廉的石墨相氮化碳催化剂仍然是一种挑战。
本文通过模板法将石墨相氮化碳(g-C3N4)的前驱体单氰胺负载在中空碳模板(hollow carbon nanospheres, HCNs)的表面,经可控热解后,获得石墨相氮化碳包覆中空碳(HCNs@g-C3N4)复合催化剂。通过扫描电镜、透射电镜、X射线衍射、红外吸收光谱对催化剂进行了一系列的形貌、结构和成分表征,并通过电化学测试研究了HCNs、g-C3N4和HCNs@g-C3N4的ORR性能。结果表明,组成复合材料后,ORR性能得到显著提高。
2. 实验部分
2.1. 试剂
硫脲(CH4N2S,>99.0%)、单氰胺(CH2N2,50%水溶液,含0.1%甲酸稳定剂)、吡咯(C4H5N, CP)、苯乙烯(C8H8,>99.5%)和十二烷基硫酸钠(SDS,>99.0%)购自阿拉丁化学有限公司。吡咯和苯乙烯单体在使用前进行减压蒸馏。碳酸钠(Na2CO3,>98.0%)、过硫酸钾(K2S2O8,>99.0%)、过硫酸钠((NH4)2S2O8,>98.0%)购自国药化学股份有限公司。氢氧化钾(KOH,>95.0%)、乙醇(C2H5OH)。除了吡咯和苯乙烯之外,所有化学品和试剂在使用前都没有进一步纯化。
2.2. 实验过程
2.2.1. 中空碳纳米球的制备
聚苯乙烯纳米微球:根据以前报道的方法 [18]:将无水碳酸钠(0.1 g, 0.94 mmoL)和十二烷基硫酸钠(0.2 g, 0.69 mmoL)溶解在300 mL蒸馏水中,然后转移到圆底烧瓶中,N2鼓气30分钟。随后,快速加入苯乙烯(30 mL),在60℃下强烈搅拌30分钟。最后,将K2S2O8水溶液(10 mL, 1 mmoL)倒入上述反应体系中,加热至75℃,并保持20小时。冷却至室温后,用蒸馏水以12,000 rpm/min的速度离心产物20分钟数次。将获得的聚苯乙烯纳米微球再次分散在100 mL蒸馏水中,称之为聚苯乙烯母液(polystyrene, PS母液)。
中空碳纳米球:将PS母液(2 mL)分散到含有100 mL去离子水的烧杯中,在搅拌下向上述体系中加入0.1 ml吡咯单体,随后逐滴加入过硫酸钾溶液(20 mL, 20 mmoL)进行聚合反应。聚合四小时后,过滤混合物并用去离子水和乙醇洗涤数次。将固体产物在60℃的真空烘箱中干燥一整夜,再将其置于管式炉中以5℃/min的加热速率从20℃升温至850℃,在此温度下热解两小时后自然冷却至室温,获得产物记为中空碳纳米球。
中空碳纳米球的酸化:取200 mg的中空碳球,置于25 mL空烧杯中,加入5 mL浓H2SO4,再缓慢加入10 mL浓HNO3,室温搅拌48小时后,加水稀释、抽滤,得到的产物在60℃下真空干燥一整夜,最终产物记为HCNs。
2.2.2. 石墨相氮化碳的制备
将适量硫脲置于管式炉内,以3℃/min的加热速率从20℃升温至550℃,在此温度下保温两小时后自然冷却至室温,得到的产物记为g-C3N4。
2.2.3. 石墨相氮化碳包覆中空碳的制备
HCNs@g-C3N4的制备:称取10 mg酸化后的HCNs,加入5 mL小烧杯中,同时放入搅拌子。加入1 mL去离子水,超声分散均匀。取400 mg单氰胺(50%水溶液,含0.1%甲酸稳定剂),并依据乙醇:水为3:1的比例将其稀释溶解,分两次加入小烧杯,搅拌直至液体挥发尽。将固体样品在70℃下真空干燥一夜,干燥后的固体样品放置于管式炉中,以3℃/min的加热速率从20℃升温至550℃,保温两小时后自然冷却至室温,即可获得HCNs@g-C3N4复合催化剂。
3. 结果与讨论
3.1. 催化剂形貌表征及结构分析
扫描电子显微镜(scanning electron microscope, SEM)是一种对微区形貌进行高分辨的分析手段。如图1(a)所示为g-C3N4的扫描电镜图片,其表现出大量石墨状片层结构堆积聚集的形貌特征,不过相互之间聚集较为松散。如图1(b)是对HCNs的形貌进行了表征,观察到通过乳液聚合合成的HCNs具有良好的分散状态,表面光滑,粒径尺寸保持在200 ± 10 nm。图1(c)、图1(d)是HCNs@g-C3N4复合物的扫描电镜图片,小球的表面较为粗糙,从断面可以观察到球壳具备了一定的厚度,表明经过高温热解后得到的g-C3N4包覆了一定量在球壳的表面。

Figure 1. SEM image of g-C3N4 (a), HCNs (b), HCNs@g-C3N4 (c) (d)
图1. g-C3N4 (a), HCNs (b), HCNs@g-C3N4 (c) (d)的扫描电镜图像
透射电子显微镜(transmission electron microscope, TEM)是使用电子来展示物件的内部或表面的技术设备。如图2(a)所示为g-C3N4的TEM图片,进一步表明我们制备的g-C3N4是具有高纵横比的纳米薄片,体现出良好的分散性。图2(b)所示为具有明显核壳状结构的HCNs,该结构使其具有高比表面积和高体积比。图2(c)是HCNs@g-C3N4复合物的TEM图片,在550℃高温热解条件下将单氰胺转化成g-C3N4负载在中空碳表面。从图中可以观察到球壳的厚度明显变大,表面有隐约分层并且较为粗糙,负载厚度达到10 ± 2 nm,说明g-C3N4成功的负载在氮掺杂中空碳基底表面。选取2(d)图中的HCNs@g-C3N4复合碳球对其进行元素映射,2(e)、2(f)显示碳、氮均匀的分散在整个中空碳球壳表面。从而可以分析得出,复合物整体呈现良好的包覆状态,中空碳表面的氮掺杂含量明显增加。此外,这种独特的核壳结构有利于O2进入催化剂表面,能够在氧气还原过程中促进电子的快速扩散。

Figure 2. TEM image of g-C3N4 (a), HCNs (b), HCNs@g-C3N4 (c); Element mapping representation of HCNs@g-C3N4 (d) (e) (f)
图2. g-C3N4 (a), HCNs (b), HCNs@g-C3N4 (c)的透射电镜图像;HCNs@g-C3N4 (d) (e) (f)的元素映射图像
我们进一步通过X-射线衍射仪(X-ray diffractometer, XRD)对样品的微观结构进行表征分析。如图3所示为硫脲经过高温热解形成的g-C3N4的XRD图,其只显示了碳的衍射峰位,在27.3˚处的强峰对应的是g-C3N4的(002)晶面,13.1˚的弱峰对应g-C3N4的(100)晶面(JCP-DS 87-1526) [19],该衍射峰是由于具有芳环结构的单元层间堆垛形成。图中没有显示出其他杂质峰,说明材料的纯度较高。HCNs的XRD图在24.7˚处出现强峰,在44˚左右出现弱峰,属于具有一定结晶度的无定形碳(002)和(100)晶面。两种单一样品复合之后的XRD衍射图显示,HCNs@g-C3N4整体的峰位置与HCNs的衍射峰位置大致相似,表明g-C3N4的包覆没有明显改变HCNs的理论结构,但是通过复合反应提高了其表面的石墨化程度,对改善HCNs的电催化稳定性产生了潜在的效用。
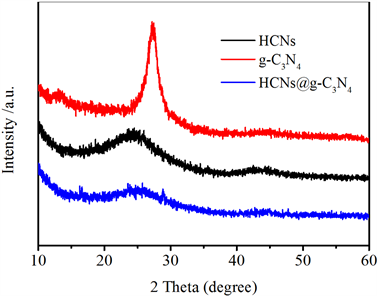
Figure 3. XRD image of HCNs, g-C3N4, HCNs@g-C3N4
图3. HCNs, g-C3N4, HCNs@g-C3N4的X-射线衍射图像
利用红外吸收光谱仪(infrared absorption spectrum, IR)对样品的结构进行分析。图4所示为HCNs、g-C3N4、HCNs@g-C3N4的红外光谱图。810.2 cm−1处对应的是嗪环单元弯曲振动峰,在1200~1600 cm−1之间显示的峰对应的是g-C3N4中的C-N和C=N的振动伸缩峰,则进一步证明合成的材料为石墨相氮化碳 [20]。在图示中,可以明显的观察到HCNs的氮掺杂程度要低于g-C3N4,与XRD图谱中的信息相吻合。将两种材料复合之后,碳氮键对应的峰位表明复合物的氮掺杂水平得到了一定的提升。
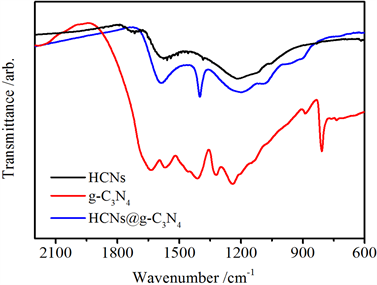
Figure 4. IR image of HCNs, g-C3N4, HCNs@g-C3N4
图4. HCNs, g-C3N4, HCNs@g-C3N4的红外光谱图像
3.2. 材料的电催化性能
在0.1 mol∙L−1的KOH碱性电解液中,采用三电极体系,在电压区间0.1 V到1.2 V (vs. RHE)上对材料的ORR电催化性能进行测试。在测试前通氧半小时,保持电解液内的氧气饱和。
HCNs、g-C3N4、HCNs@g-C3N4样品在扫描速率为50 mV∙s−1的循环伏安曲线(CV)如下图5(a)所示。从图中可以很明显的观察到阴极氧气还原峰,表明三种样品都有一定的氧气还原活性。但是HCNs@g-C3N4复合物的阴极峰电流密度更大,电位更正。测试表明,HCNs@g-C3N4复合物表现出最好的ORR催化活性。
利用旋转圆盘电极对样品的ORR催化活性进行评价。HCNs@g-C3N4在不同转速下的线性扫描伏安曲线(LSV)如图5(b)所示,从圆盘电极收集的响应电流随着转速的增加而增加,表明HCNs@g-C3N4具有优异的电子传输能力。HCNs、g-C3N4、HCNs@g-C3N4在1600 rpm下的LSV曲线如图5(c)所示,在电流密度为−0.1 mA∙cm−2时,HCNs@g-C3N4复合物的起始电位(1.087 V)比HCNs (0.770 V)、g-C3N4 (0.777 V)表现的更正。同样的,HCNs@g-C3N4复合物的半波电位也相比于单一样品更正。很显然,HCNs@g-C3N4复合物在0.3 V时的极限电流密度(3.286 mA∙cm−2)也远高于g-C3N4 (1.957 mA∙cm−2)、HCNs (2.424 mA∙cm−2)。这主要是因为两种材料复合之后形成的特殊核壳结构使得其比表面积和导电率增大,有利于相表面的物质传输和电子转移,使得ORR性能得到有效提升。

Figure 5. (a) CV curves of HCNS, g-C3N4, HCNs@g-C3N4 at the scanning rate of 50 mV∙s−1; (b) LSV curves of HCNs@g-C3N4 at different rotating speeds; (c) LSV curve of HCNS, g-C3N4, HCNs@g-C3N4 at 1600 rpm; (d) I-T curve of HCNS, g-C3N4, HCNs@g-C3N4
图5. (a) HCNS, g-C3N4, HCNs@g-C3N4在50 mV∙s−1的扫描速率下的CV曲线;(b) HCNs@g-C3N4在不同转速下的LSV曲线;(c) HCNs, g-C3N4, HCNs@g-C3N4在1600 rpm下的LSV曲线;(d) HCNS, g-C3N4, HCNs@g-C3N4的I-T曲线
采用10,000 s恒电位测试,进一步评价样品的电催化稳定性能,I-T曲线如图5(d)所示。其中g-C3N4表现出的循环稳定性最好,在10,000 s后电流密度达到102.3%,得益于类石墨的片层状稳定结构,而HCNs的电流密度仅仅是原来的72.3 %。将两种材料复合之后,在ORR催化性能大幅度提升的基础上,也使得稳定性达到了良好的综合,保持了原有电流密度的86.2%。在恒电位测试中,复合物的催化活性损失的较轻微,表明HCNs@g-C3N4复合物催化剂上的活性位点在碱性介质中较为稳定。
4. 结论
本文的创新在于将g-C3N4包覆在导电基底中空碳表面。g-C3N4的含氮量高,对O2的吸附活化性能较好。HCNs@g-C3N4复合催化剂的制备克服了其导电性能不好的缺点。另外,HCNs@g-C3N4独特的三维核壳状结构,综合了两相的优点,表现出良好的ORR催化活性及稳定性。在后续的工作中,我们可以进一步在此复合催化剂表面负载金属或者非金属纳米粒子,为制备更加优异的ORR催化剂提供了坚实的基础。
基金项目
这项工作受江苏省高等学校自然科学研究面上项目(20KJB430046)和南通大学科研启动金(03083030)支持。
参考文献