1. 引言
随着手性药物在新药中所占比重逐年增大,不对称催化也面临着越来越多的发展机遇与挑战。烯丙基化合物是一种重要的有机合成中间体,许多活性天然产物结构中含有烯丙基片段。经过几十年的发展,不对称烯丙基化反应已成功应用于多种类型手性化合物的合成,并在合成生物分子、药物、天然药物等多方面得到广泛应用。过渡金属催化的不对称烯丙基化反应以其高度区域和立体选择性等优点成为近年来的研究热点,也是构筑碳–碳键和碳–杂键的重要方法之一 [1] [2] [3] 。由于活性的烯丙基试剂参与的烯丙基化反应会生成较多的副产物,且会造成原料的浪费,因此,近年来,惰性烯丙基试剂开始广泛应用于烯丙基化反应中,很好地解决了活性烯丙基试剂参与反应的不足 [4] 。经过大量的实验证明,惰性烯丙基试剂如烯丙基醇、烯丙基醚、烯丙基胺等具有如下优点:1) 不易产生副产物;2) 简化反应步骤;3) 通常较稳定,可存在于某些天然产物以及一些重要的化学中间体中。由于烷基胺、烷氧基、羟基很难离去,因此直接将烯丙胺 [5] 、烯丙醚和烯丙醇 [6] 作为烯丙基试剂用于不对称烯丙基化反应仍然是一个挑战。
2. 惰性烯丙基试剂参与的不对称烯丙基化反应
2.1. 烯丙基醇作为烯丙基前体
相比于烯丙基酯试剂,烯丙基醇作为亲电试剂具有以下几个优点:一是来源广泛,简单易得;二是该反应副产物多数都为水,原子经济且环境友好。因此,烯丙基醇试剂常作为亲电试剂被广泛应用于不对称烯丙基化反应中。
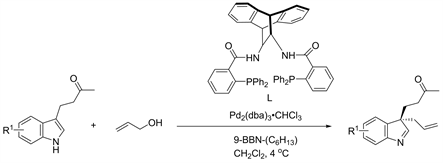
Figure 1. C-3 allylation of 3-substituted-1H-indoles
图1. 3-取代-1H-吲哚的C-3位烯丙基化反应
2006年,Trost课题组报道了吲哚C-3位的烯丙基取代反应。9-BBN-(C6H13)作为活化的硼试剂,通过加入手性配体(L*)以实现并提高反应的对映选择性 [7] (图1)。
2013年,龚流柱课题组 [8] 首次通过金属钯/手性磷酰胺与手性磷酸协同催化,实现了烯丙醇和吡唑啉酮的不对称烯丙基化反应(图2)。在最佳配体和最优反应条件下,对带有不同取代基团的底物进行适用性探究,最终得到一系列具有全碳季碳手性中心的多取代吡唑啉酮烯丙基化产物,且产物具有优异的产率(高达99%)以及良好的对映选择性(高达97% ee)。

Figure 2. Asymmetric allylic alkylation of pyrazol-5-ones with allylic alcohols
图2. 吡唑酮与烯丙醇的不对称烯丙基化反应
针对良好的对映选择性实验结果,该课题组提出一个可能的反应机理(图3)。手性磷酸(PA)通过氢键作用活化烯丙醇形成络合物B,在钯作用下C-O键断裂形成π-烯丙基钯中间体C,同时脱去一分子H2O。吡唑啉酮发生烯醇互变,与手性磷酸通过氢键作用,立体选择性地控制中间体C的亲核取代过程。反应的决速步中,通过手性磷酸根与π-烯丙基钯络合物之间的协同作用,对反应的立体选择性进行了很好的控制,得到烯丙基化目标产物并完成催化循环。反应中,通过Pd/L*与手性PA协同催化,实现了高效、高立体选择性的烯丙基化反应。

Figure 3. Proposed catalytic cycle for allylic alkylation of allylic alcohols with pyrazol-5-ones
图3. 烯丙基醇与吡唑啉酮反应可能的催化循环
2014年,蒋高喜和夏春谷课题组在前人工作的基础上,以Brønsted酸作为促进剂,在Pd催化作用下实现了吖内酯与烯丙基醇的不对称烯丙基化反应 [9] ,反应最终得到了一系列直链的烯丙基化产物,提供了一种新型高效的合成手性氨基酸类衍生物的方法(图4)。与前人工作相比,在该反应中,全程没有碱的参与,所得副产物为水,且为原子经济性反应。该反应首先采用带有不同供电子和吸电子取代基的吖内酯和烯丙基醇进行反应,得到具有良好对映选择性的目标产物(ee值达90%)。在对带有不同取代基的直链及支链烯丙醇参与的反应进行研究时发现,只有在得到直链产物的反应过程中,才有π-烯丙基钯中间体的生成从而对反应起到促进作用。

Figure 4. Asymmetric allylation of azlactones with cinnamyl alcohols
图4. 吖内酯与烯丙基醇的反应
2.2. 烯丙基胺作为烯丙基前体
烯丙基胺试剂同样具有原料易得、参与反应为原子经济性反应的优势,因此烯丙基胺也受到广泛关注。Pd催化的烯丙基胺参与的不对称烯丙基化反应为含烯丙基底物的合成提供了另一种可行的方法。由于C-N键的稳定性,涉及到烯丙基胺的系列反应具有挑战性以及极高的可研究价值。
借助含氮杂环的环张力可促进烯丙胺参与的烯丙基化反应,如乙烯基-氮杂环丙烷能够被用于烯丙基的取代反应中。2007年,Trost课题组使用Trost双膦配体以及[Pd(η3-C3H5)Cl]2共同组成的催化体系进行催化,实现了乙烯基氮杂环丙烷与二酰亚胺的动态动力学不对称烯丙基胺化反应。在该反应中,发生了酰基迁移从而得到N1-酰基-N2-叔丁氧羰基-邻二胺,且产物具有较高的产率以及良好的对映选择性 [10] (图5)。

Figure 5. [Pd(η3-C3H5)Cl]2/L* catalyzed asymmetric allylicamination
图5. [Pd(η3-C3H5)Cl]2/L*催化的不对称烯丙基胺化反应
2011年,张万斌课题组 [11] 报道了羰基化合物与多种烯丙基胺在Pd/L*催化条件下通过氢键活化所发生的烯丙基化反应。该反应通过Pd与烯丙基胺作用促进C-N键断裂,使烯丙基与羰基碳作用形成新的C-C键(图6)。该方法拓展应用到多种烯丙基胺及多种羰基化合物底物中,均得到较高的产率以及良好的对映选择性。由于该反应中使用甲醇作为溶剂,因此该反应为基于Pd催化的烯丙基胺的烷基化反应来形成新的C-C键的反应提供了一种更为经济便利的方法。

Figure 6. Allylic alkylation of allylic amines with carbonyl compounds
图6. 烯丙基胺与羰基化合物的烯丙基化反应
2012年,田仕凯等人 [12] 使用B(OH)3作为活化剂,进行了亚磺酸盐与烯丙基伯胺的烯丙基取代反应。在该反应中,烯丙基伯胺中的-NH2基团直接被亚磺酸盐取代,反应具有良好的立体和区域选择性。烯丙基砜类化合物可以通过烯丙基伯胺在BINOL配体的作用下与亚磺酸盐作用而制得(图7)。

Figure 7. Direct substitution of primary allylic amines with sodium sulfinates
图7. 烯丙基伯胺与亚磺酸盐的直接取代反应
同年,该课题组报道了Pd催化的α-手性烯丙基伯胺与芳基硼酸的立体专一的交叉偶联反应。反应所得偶联产物具有良好的对映选择性 [13] (图8)。

Figure 8. Cross coupling of α-chiral primary allylic amines with aryl boronic acids
图8. α-手性烯丙基伯胺与芳基硼酸的交叉偶联反应
2016年,田仕凯课题组 [14] 报道了关于Pd催化的硝基乙酸盐与烯丙基伯胺的立体专一的烯丙基化反应(图9)。

Figure 9. Stereo specific allylation and decarboxylation/denitronation
图9. 烯丙基化反应和脱羧反应/去硝基化
在该反应中,在Pd催化剂和硼酸同时存在的条件下,通过一锅法发生脱羧反应,实现一系列硝基乙酸盐与烯丙基伯胺的烯丙基化反应,从而得到多种高烯丙基硝基化合物,且产物具有较高产率(图10)。同时,该反应伴随着去硝基化的发生,为手性β-取代-γ,δ-不饱和酯类化合物的合成提供了路线。
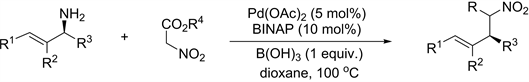
Figure 10. Allylation of nitroacetates with enantioenriched primary allylic amines
图10. 硝基乙酸盐与烯丙基伯胺的烯丙基化反应
2.3. 烯丙基醚作为烯丙基前体
烯丙基醚,特别是烯丙基烷基醚参与的取代反应是目前最具有挑战性的烯丙基取代反应之一。在过渡金属催化的不对称烯丙基取代反应中,为了提高反应活性,烯丙基醚试剂经常被用作烯丙基环氧化合物。由于其稳定性,在烯丙基醚参与的过渡金属催化的烯丙基取代反应中经常需要使用路易斯酸或者强亲核性的格氏试剂作为活化剂。
2014年,张万斌课题组 [15] 报道了由[Pd(η3-allyl)Cl]2、dppf以及四氢吡咯的甲醇溶液所组成的催化体系用于烯丙基烷基醚的氢键活化作用中。其中,在Pd催化下,烯丙基烷基醚的C-O键的断裂,该反应中溶剂醇发生氢键活化是实现烯丙基烷基化反应的关键(图11)。该反应不需要使用任何添加剂且具有良好的区域选择性。同时,该课题组对不同取代的烯丙基醚底物参与反应的普适性进行了研究,发现线型烯丙基醚比带有支链的烯丙基醚反应活性更高。

Figure 11. C-O bond cleavage of allylic alkyl ether
图11. 烯丙基醚的C-O键断裂
2.4. 联烯作为烯丙基前体
联烯类化合物也可以作为烯丙基化试剂,不需要额外离去基团,反应过程不产生任何废料,有很好的原子经济性。
2017年,Breit课题组 [16] 在金属铑的催化下,手性亚磷酰胺作为配体,联烯作为烯丙基前体,成功实现了β-二酮的不对称烯丙基化反应。反应中加入的手性配体与添加剂TFA共同作用,对于亲核试剂高选择性地进攻π-烯丙基铑中间体起到很好的调控作用,从而高产率(99%)、高对映选择性(96% ee)的得到烯丙位取代的端烯产物(图12)。
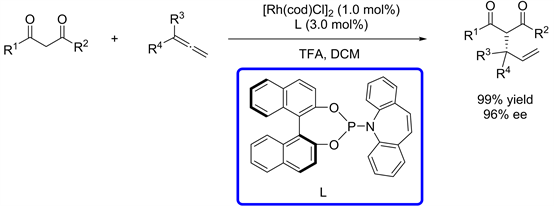
Figure 12. Rh-catalyzed asymmetric allylation with allene
图12. 铑催化的联烯参与的不对称烯丙基化反应
2017年,蒋高喜课题组 [17] 设计并实现了联烯醚作为烯丙基前体与吡唑啉酮的高区域和高立体选择性加成反应(图13)。在该反应中,一种途径是通过金属钯进行催化,得到吡唑啉酮的4位进行支链烯丙基化加成产物;另一途径是通过手性磷酸催化,得到吡唑啉酮的4位进行直连烯丙基化加成产物。

Figure 13. Asymmetric allylation reaction involving allene ether
图13. 联烯醚参与的不对称烯丙基化反应
2018年,Buchwald课题组 [18] 报道了一种高对映选择性Cu催化的联烯参与的烯丙基化反应(图14)。该反应使用酮与不同官能团的联烯进行偶联,得到一系列高选择性的支链烯丙基化产物。

Figure 14. Asymmetric allylation of allene and ketone
图14. 联烯与酮的不对称烯丙基化反应
3. 总结
综上所述,近年来,通过多种类型的惰性烯丙基试剂作为烯丙基前体所参与的不对称烯丙基化反应已经得到了较快的发展。烯丙基醇、胺、醚类化合物在该类反应中的应用,为碳碳键及碳杂键的构筑提供了新的研究途径,且更为经济环保。
尽管这些方法迅速发展,但也存在一定的难点和挑战:1) 用烯丙醇进行高效且高度立体选择性的烯丙基取代反应仍然相对具有挑战性。虽然手性配体和添加剂已经有了一定的发展,但底物的作用范围是十分有限的。2) 涉及烯丙基醚、胺的大多数反应通常需要使用乙烯基环氧化物和乙烯基氮杂环丙烷类化合物,以及相对苛刻的反应条件。3) 许多该类烯丙基取代反应仍然需要使用添加剂(如路易斯和质子酸)来进行活化。
尽管上述挑战众多,但烯丙基取代反应的最新进展表明,它们并非不可克服。这些困难为科研工作者们提供了一个很好的机会,使该领域取得更好更快的发展。
基金项目
国家自然基金项目(No. 21462042)。
NOTES
*通讯作者。