1. 引言
质子转移可能是最基本的化学反应过程,当中性有机分子转化为带电荷的共轭酸或碱形式时,会导致中性有机分子的反应性大大提高。因此,人们对能够探测质子转移过程范围的方法普遍很有兴趣。溶剂是化学实验室的重要和必要成分。由于溶剂改变了与分离的分子相比的分子特性,因此理解溶剂效应是重要的,且理解和合理化溶剂和液体环境对分子性质的作用的理论发展是非常重要的。近年来,在这方面投入了大量的理论努力。为了解解决方案中的化学反应,仍然需要进一步开发。
Daniela课题组 [1] 以DFT/B3LYP6-311G**理论水平分析并表征了甲酰胺和硫代甲酰胺中分子内质子转移反应的机理以及水分子在化学过程中的作用。他们通过分析能量,反作用力,反应电子通量,电子种群以及分子中原子的Bader量子理论对系统进行了全面表征。并且,他们发现PT方法在热力学上是不利的,因为在所有情况下氨基形式都更稳定,而反作用力分析有助于量化电子和结构对能垒的贡献。在所有情况下,结构重排都是主要的。另一方面,已经表明水分子起催化剂的作用。在两个反应中,能垒均降低了两倍。
张课题组在研究2,3,5-三甲基苯醌加氢催化的反应中,通过利用理论计算方法以及真实实验操作相结合,得出最终结果进行比较 [2]。通过各种技术进一步表征制备的催化剂。如图1所示,NH2-UiO-66和2.0 wt% Pd@NH2-UiO-66的XRD图谱与从晶体结构数据模拟得出的UiO-66几乎相同。在当前的动态条件下可以很好地建立NH2-UiO-66,其中添加Pd前体不会影响NH2-UiO-66的生长,因此在DMF于130%还原处理后其框架保持完整。此外,没有观察到描述Pd纳米颗粒的峰,表明封装于MOF基质内的超小型Pd纳米颗粒的低负载和高分散。选用此种的催化剂的原因在于其具有较好的催化性能以及良好的热稳定性 [3] [4]。

Figure 1. XRD patterns of UiO-66 simulated from crystal structure data, the parent NH2-UiO-66, and the one-pot synthesized 2.0 wt% Pd@NH2-UiO-66
图1. 由晶体结构数据模拟的UiO-66的XRD图谱,母体NH2-UiO-66和一锅法合成的2.0 wt% Pd@NH2-UiO-66 [2]
在理论计算过程中,基于色散矫正密度泛函理论(DFT)的方法用于研究使用包封Pd团簇的NH2-UiO-66氢化TMBQ的机理。用PBE的广义梯度近似函数用于描述交换函数。使用周期性晶体结构代替团簇模型来再现纳米笼对反应物吸附的限制作用。脱水NH2-UiO-66的原始细胞含有120个原子,只有伽马点用于取样布里渊区。移动所有优化的结构,直到每个原子的绝对力小于0.03 eV/Å。采用爬山图像轻推弹性带法检测TMBQ氢化过程中的过渡态,确定所得过渡态只有一个虚频。所有计算均使用VASP进行。
结论中提出了两步反应用来说明TMBQ氢化的反应机理,即第一和第二步反应步骤分别代表第一个酮基和另一个酮基(对位)的氢化。NEB计算表明,第一反应步骤的能垒为80.7 kJ/mol。在第一步反应的最终状态(FS1)中,dH-O为0.99 Å,质子与Pd原子之间的键断裂,距离为2.36 Å,此步骤略微吸热,能量为10.1 kJ/mol。发现第二步反应的初始状态IS2与IS1相比具有较低的能量−40.2 kJ/mol,并且NEB计算表明第二步反应的能垒小得多,即34.5 kJ/mol。TS2结构中质子与来自第二个酮基的O原子之间的距离为1.60 Å,类似于TS1中的1.67 Å。在第二反应的最终状态(FS2)中,两个酮基团的氢化结束,并且TMBQ产物物理吸附在八面体笼中。第二步的反应能垒小得多,其放热效应表明在TMBQ氢化过程中产生的中间体(即FS1)可以很容易地转移到产物中,这与TMBQ的氢化可以达到高产率的实验现象非常吻合。
当然,此研究的计算结果从原子水平上对反应机理进行了很好的阐述。但是从数据结果比较来看,理论计算出来的活化能值为80.2 kJ/mol,而实验测出来的值为36.4 ± 0.3 kJ/mol。所以从中可以看出理论值与实验值还是有一定的差距,说明在催化反应中考虑水溶剂效应具有十分重要的意义。而为此,我认为可以提出的解决方式主要有以下两种。一是直接考虑溶剂化模型进行计算;二是采用真实的水模型重新对反应体系进行机理研究,从而判断水分子参与的情况是否对质子转移有明显的影响作用。由于目前为止,关于此类的水模型建立并研究计算并不成熟,我们的研究也处在摸索之中。
2. 理论计算方法
2.1. 分子动力学
在水溶液对8-羟基鸟嘌呤自由基水辅助质子转移机理的研究中,利用组合量子力学和分子力学理论 [5] [6] [7],研究了8-OHGrad在水中的水辅助分子内质子转移,其中具有一个水分子的8-OHGrad复合物被处理为具有DFT和有效静电势(ESP)的QM区域。在不同计算阶段的理论水平,水溶液被视为经典的MM区域。将8-OHGrad∙H2O复合物包埋在39.9 Å立方体盒中,该盒由2137个水分子组成,用明确的SPC/E水模型描述 [8]。因此,利用DFT/M08-SO/cc-pVTZ+理论水平用于处理QM区域以计算沿反应途径的PMFs。此外,溶质通过键合相互作用,静电相互作用和范德华相互作用与水分子相互作用,QM区域的范德华参数从标准琥珀力场获得 [9]。PMF的计算使用NWChem计算化学包进行。使用分子动力学模拟使溶剂在整个反应途径中平衡120 ps,然后优化整个NEB反应途径。重复最后一步直至最终的NEB反应途径收敛。
2.2. 密度泛函理论
而在蝶呤的水辅助质子转移反应中,使用密度泛函理论(DFT)方法B3LYP [9] /6-31+G(d, p) [10] [11],和PBE [12] /6-31+G(d, p)和二阶MP2/aug-cc-pVDZ理论水平优化基本形式(B),酸形式(A)和过渡态(TS)的2-氨基喋呤-4(3H)-一种蝶呤的几何结构。通过使用谐波近似计算振动频率来确认静止点的性质。确认了四种结构的真实最小值:A,B,AW和BW。转换状态TS和TSW也仅用一个假想频率确认。频率计算还提供了能量的自由能和相关贡献,例如室温下的零点振动和热校正。
3. 质子转移反应机理
3.1. 双质子转移
溶剂还可以通过直接参与化学反应来改变气相反应机理。例如,如图2所示,对于甲酰胺与甲脒酸反应的互变异构化,发现水分子有助于甲酰胺在水溶液中的互变异构,并且与气相中的相比降低了阻隔高度。另一项关于OH− + CCl4在水溶液中的水辅助SN2反应的研究发现,水分子通过质子转移过程有助于在有利的背面攻击构象中形成新的OH−然后新形成的OH−攻击基板CCl4。此外,溶剂可以同时用作溶剂和催化剂。
水辅助质子转移过程是水溶液中羟基自由基对鸟嘌呤损伤反应的关键步骤。绘制的原子级反应途径,显示了辅助水分子与8-羟基鸟嘌呤基团之间的同步双质子转移机制。过渡态偶极矩是沿反应路径最大的,其静电稳定质子转移过渡态复合物。
在与RNA和DNA碱基的反应中观察到水辅助的质子转移机制,包括尿嘧啶,胞嘧啶,胸腺嘧啶,腺嘌呤和鸟嘌呤。对于一水合鸟嘌呤复合物的分子内质子转移的量子化学研究发现,与非水辅助过程相比,互变异位氧–氢反应的反应势垒高度约低两倍。密度泛函理论(DFT)的从头算研究和C8氧化鸟嘌呤中分子内质子转移理论的MP2水平表明,水分子在质子转移过程中的辅助大大降低了反应势垒。使用DFT和连续溶剂模型(积分方程形式–极化连续模型(IEF-PCM))的第一性原理研究发现,8-氧代-7,8-脱氢嘌呤中的互变异构化的水分子辅助质子转移过程可以在水溶液中显着降低活化吉布斯能量至小于10 kcal/mol。对于8-羟基鸟嘌呤基团(8-OHGrad),从头算多组分MCSCF/DZP/MRPT2研究表明,气相中水辅助质子转移的活化势垒为22.4 kcal/mol [13],比没有一个水分子辅助的直接反应过程约小于26.3 kcal/mol。在水溶液中,具有IEF可极化连续溶剂化模型的DFT/B3LYP理论发现8-OHGrad与一个水分子的水合质子转移为18.6 kcal/mol。尽管作者描述了沿着水分质子转移过程的反应途径的静止点,但反应途径的详细的原子级演变是不可行的,溶剂效应和溶剂对反应屏障的定量贡献是还是一个未知数。
质子转移过程必须通过第一和第二溶剂化壳的重组中水分子的偶极矩分布来辅助;因此,正确地再现水的微观重排分子,应该明确地处理溶液水分子。然而,到目前为止,还没有关于8-OHGrad水溶液中水辅助质子转移的研究,以分析溶剂效应对反应途径中能量学的定量贡献;因此,水溶液对活化屏障的催化作用是未知的。此外,还没有详细的反应途径。
Figure 2. Schematic diagram of the mechanism structure change of the reaction path
图2. 反应路径机理结构变化示意图 [14]
3.2. 互变异构
蝶呤的水辅助质子转移反应中,要考虑的一个重要方面是溶剂的溶质极化。溶质极化是主要关注的主题,并且已经使用不同的程序包括在内。这里的溶质极化是参考分子的静电力矩的变化,这是由溶剂提供的电场的结果。为了解释这一点,科研人员使用了迭代程序,在溶剂环境中静电平衡溶质。在水辅助机制中,我们考虑另外一个水分子作为溶质的一部分,如图3所示。在MP2/aug-ccpVDZ水平下,酸

Figure 3. Optimized structures of pterin in the intramolecular process of tautomerization. B is the basic form, TS is the transition state, and A is the acid form. The distances shown were obtained from the MP2/aug-cc-pVDZ geometry optimization
图3. 水辅助互变异构化过程中蝶呤的优化结构。BW是基本形式,TSW是过渡态,AW是酸形式。全部用一个水分子优化,显示的距离是从MP2/aug-cc pVDZ几何优化中获得的 [15]
性互变异构体的气相偶极矩(在此被认为是A)为3.68D,在水环境中增加至5.50D,增加约50%。酸互变异构体A和AW显示出比碱性互变异构体更大的偶极矩,因此表明在水溶液中酸形式与水的相互作用更强,并且溶剂在增加酸形式的稳定性中起重要作用。
对于水辅助机制,水环境使过渡状态更稳定(−3.70 kcal/mol),将隔离分子的无障碍能量从10.37 kcal/mol变为溶剂化状态的6.67 kcal/mol。酸性互变异构体稳定在−7.96 kcal/mol,相对自由能从−3.25变化到−11.21 kcal/mol。因此,碱性酸互变异构体之间的溶剂化差异自由能为−11.21 kcal/mol,其中−7.28 kcal/mol来自静电项,−3.93 kcal/mol来自其余项,因为几何变化和范德华互动。图4总结了包括两种机制中质子转移中水性环境的影响的结果。它还表明在水辅助机制中活化自由能相当低。在水溶液中的水辅助机制中,分子内机制中的32.55 kcal/mol的自由能垒降低至仅6.67 kcal/mol。它表明水分子直接参与反应和水的体积效应降低了25.88 kcal/mol的阻隔,增加了反应速度。在酸性互变异构体的稳定化中观察到水的相反作用。在分子内机理中,它比碱性互变异构体稳定16.36 kcal/mol,在溶液中的水辅助机理中降至11.21 kcal/mol;它是5.15 kcal/mol的不稳定。
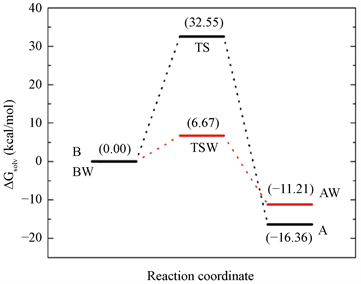
Figure 4. Schematic free energy profile for both tautomerization processes in solution, intramolecular and water-assisted
图4. 溶液,分子内和水辅助互变异构过程的示意图自由能谱 [15]
研究中都有水分子直接参与了反应,但是主要涉及的是分子内的异构化,反应物分子与水分子形成了稳定的中间体,降低了反应活化能,促进反应的发生。
4. 水溶剂效应
水溶液中羟基自由基对鸟嘌呤损伤反应中,溶剂效应能对扭转角度产生巨大影响,远远大于弯曲角度和粘合距离。不仅是角度,还有水溶液中的键长。然而,目前的反应体系具有致密的环结构,并且溶剂效应不足以影响静止点中的强键相互作用,而较弱的扭转相互作用可能容易受溶剂效应影响,特别是通过氢键在它们的氢壳中形成。因此,对于当前系统,静止点构象的最大差异在于它们在气相和溶液相之间的相应扭转角。
过渡态偶极矩是沿反应路径最大的,其静电稳定质子转移过渡态复合物。利用密度泛函理论/M08-SO/cc-pVTZ+/分子力学理论水平,以19.2 kcal/mol计算该水合质子转移反应的自由能反应势垒。溶剂效应不仅对几何形状有很大影响,而且还会显着改变反应途径的能量。在溶剂效应对过渡态的贡献中,溶剂能量贡献为−28.5 kcal/mol,极化效应贡献为19.9 kcal/mol。总之,溶剂效应对自由能垒高度贡献−8.6 kcal/mol,这意味着水溶液的存在对反应机理具有催化作用并且增强水溶液中的质子转移反应性。
蝶呤是存在于多种生物系统中的杂环化合物家族的成员,并且可以以两种形式存在,对应于酸和碱性互变异构体。在这项工作中,在气相和水溶液中研究了这些互变异构形式之间的质子转移反应。在气相中,通过量子力学二阶Møller-Plesset扰动理论(MP2/aug-cc-pVDZ)计算对分离的蝶呤进行分子内机制,表明酸形式比基本形式更稳定,−1.4 kcal/mol,相对于基本形式具有34.2 kcal/mol的阻隔。在水溶液中,水分子在质子转移反应中的作用在两个分开的部分中分析,一个水分子直接参与反应路径,称为水辅助机制,以及水溶剂化的互补参与。通过量子力学计算对一个蝶呤水簇进行了水辅助机理,表明酸形式仍然更稳定−3.3 kcal/mol,同时屏障的70%急剧减少。蒙特卡罗模拟中实现的自由能扰动包括对分子内和水分机制的大量溶液效应。当反应路径涉及水辅助机制时,发现大量水效应是实质性的和决定性的。
除此之外,Dong课题组 [16] 利用DFT计算方法研究水助Al3+在水溶液中自发水解的协同质子离解机理。在这项研究中,我们了解到利用密度泛函理论计算还可以阐明第二级壳水辅助的协同质子解离机理,这是作为水溶液中Al3+一级自发水解的主要机理。反应途径通过两个质子从两个氢键结合的内壳和第二壳水的协同解离而进行。
从这些研究中,我们都看到了水溶剂效应对于探究化学反应机理的重要性,而我们还存在很大的探究空间。
5. 总结和展望
鸟嘌呤损伤环境中,结合量子力学和分子力学理论,研究了8-OHGrad在水溶液中的水辅助分子内质子转移。使用明确的SPC/E溶剂化模型来描述水溶液以描述水分子的局部重排。与气相中相应的静止点相比,静止点表明水溶液对二面角的影响最大。沿反应途径的详细的原子反应机理表明,反应机理是同步的双原位转移过程。在DFT/M08-SO/cc-pVTZ+理论水平下,计算的自由能垒为19.2 kcal/mol。最强的静电相互作用稳定了过渡态的质子转移过程;因此,它降低了水溶液中的反应势垒高度。计算还表明,溶剂能量对过渡态贡献−28.5 kcal/mol,极化效应贡献19.9 kcal/mol。总之,水溶液对活化屏障贡献−8.6 kcal/mol,这意味着水溶液降低了水溶液中的过渡态并且对该质子转移反应机理具有催化作用。蝶呤的水辅助质子转移过程在这项工作中考虑了两种不同的反应机制,即直接分子内质子转移和溶剂辅助机制。在气相中,MP2/aug-cc-pVDZ水平的分子内反应机理导致34.19 kcal/mol的能垒,通过三中心过渡态。与一种水分子协同的互变异构反应将能垒降低至10.37 kcal/mol,与直接机制相比,显著降低了近70%。因此,即使不考虑水环境的大量影响,水辅助机制对于获得现实屏障也是至关重要的。在室温下考虑水环境时,情况更加清晰。
综上,在研究各类催化反应时,水溶剂效应都是不可忽略的重要因素,所以对于此方向的研究也具有重要的意义。然而,目前为止,关于此类的研究还不够成熟,不够多。而我的研究方向,也将建立在此基础上,选择合适的反应体系,并建立科学的水溶剂模型,及运用合理高效的理论计算方法,对水溶液中的催化反应机理进行更深入的研究。在研究过程中,要及时将气相反应与水溶液中反应进行对比,将探索型简易水模型以及模拟真实的水溶液模型进行比较。结合一定的实验,将实验值与理论值进行比较,进一步验证及说明。
NOTES
*通讯作者。