1. 引言
pH是生物体的生理活动中的一个重要参数,在细胞增值、凋亡、酶活性和蛋白质降解等生理过程中起着关键性的作用 [1] [2] [3] [4]。典型的ESIPT荧光团2-(2'-羟基苯基)苯并噻唑、2-(2'-羟基苯基)苯并恶唑、2-(2'-羟基苯基)苯并咪唑表现出pH依赖性,在不同pH条件下,会产生阴离子和阳离子,并发出复杂的荧光 [5] [6] [7] [8] [9]。Sahu曾报道2-(2'-羟基苯基)苯并咪唑在非极性溶剂环己烷和极性溶剂乙腈中同时存在顺式醇和反式醇 [10]。2016年,Saugata [11] 等通过实验结合理论计算发现2-(2'-羟基苯基)苯并咪唑可以由中性分子转化为顺式阴离子和二价阴离子。Minati [12] 及其他的同事以2-(4'-二乙氨基-2'-羟基苯基)-1H-咪唑-[4,5-b]吡啶(DHP)为例,使用紫外–可见吸收和荧光发射光谱,结合量子力学计算探究了DHP在哈米特酸度(Hammett’s acidity scale, H0) (-10)到雅吉尔碱度(Yagil’s basicity scale, H-)) (16.4)范围内的离子结构,其研究结果表明DHP在纯溶剂以中性分子的结构存在,其中的发射带位于455 nm处。当pH = 12时,先脱去“OH”上的氢,形成一价阴离子Monoanion,当溶液的pH增大至16.2时,再脱去“NH”上的氢,形成二价阴离子Dianion,伴随着375 nm处的吸收带的生成,在466 nm处发射荧光。同样的,在酸性环境中,当pH在3.0~7范围内,咪唑环上的氮先质子化形成Monocation,当溶液的酸性由pH = 3.0减小至pH = −0.6时吡啶上的氮同样被质子化形成Dication,由于共轭程度增大,光谱均发生红移 [13] [14] [15],当溶液的pH小于−0.6时,形成Trication物种。由于DHP在酸碱性环境中的阴阳离子较宽波长范围(340 nm~685 nm)的发射,因此DHP可以做pH指示剂,指示水环境中的pH变化。不仅酸碱性可以调控其阴阳离子的产生,改变溶剂的极性,也可以调控离子的生成。
本论文选取2-(2'-羟基-5'-硝基)苯基苯并咪唑(HBI-pNO2)为研究对象。借助不同溶剂中的紫外吸收光谱,探究溶剂极性及其酸碱性对其分子的基态结构的调控,并结合理论计算对其阴离子进行结构指认。
2. 实验试剂与理论计算方法
2.1. 实验试剂
2-(2'-羟基-5'-硝基)苯基苯并咪唑(HBI-pNO2);苯、四氢呋喃、乙腈及甲醇均为市售色谱纯试剂。四丁基氢氧化氨、三氟乙酸均购买于米克。
2.2. 实验仪器
紫外分光光度计,UV-2501PC,Shimadzu Corp.,Japan。
2.3. 理论计算方法
密度泛函理论(Density Functional Theory),简称DFT [16] 是计算化学领域最常用到的方法之一,DFT的理论基础是Hohenberg-Kohn理论,该定理指出基态电子密度函数决定了系统所有的电子性质,它是从上世纪在Thomas-Fermi模型 [17] 的基础上发展起来的一种研究多电子结构的量子理论计算方法。
含时密度泛函理论(TD-DFT)是目前较为成熟的理论,它是基于Runge-Gross理论,含时电子密度ρ(r, t)确定了与时间有关的电子性质 [18]。在本论文中,使用了TD-DFT方法,B3LYP泛函和6-311 + G**基组计算了分子激发态的电子跃迁能,振子强度及轨道跃迁类型。
3. 结果与讨论
3.1. 不同溶剂中的紫外吸收光谱
2-(2'-羟基-5'-硝基)苯基苯并咪唑(HBI-pNO2)在C6H6,THF,CH3CN和CH3OH中的紫外吸收光谱,如图1所示。从图中可以看出,HBI-pNO2在C6H6和THF中,有三个吸收带,在C6H6中最大吸收波长分别位于291.2,319.2和332.0 nm处,而在THF中均略有蓝移。HBI-pNO2在极性较大的溶剂CH3CN和CH3OH中,在400 nm附近观察到一个相对较弱的新吸收带。

Figure 1. The normalized absorption spectra of HBI-pNO2 in different solvents
图1. HBI-pNO2在不同溶剂中归一化的紫外吸收光谱
为解释实验光谱,我们在B3LYP/6-311 + G(d, p)水平上优化了HBI-pNO2可能存在的五种分子构型如图2所示,分别为顺式醇(enol-1),反式醇(enol-2),enol-3,enol-4和酮(keto),从优化的键长可以看到:与enol-1相比,enol-2的N-H的键长由1.008 Å拉长至1.009 Å,O-H的键长由1.006 Å缩短至0.966 Å;扭转的构型enol-3的O-H的键长由1.006 Å缩短至1.000 Å,N···H的键长由1.658 Å增加至1.680 Å,O-H…N的分子内氢键增强,且硝基的N-O键也由1.231和1.232 Å拉长至1.224 Å;enol-4中苯并咪唑环和苯酚环由enol-1的平面扭转为非平面,且N-H的键长变长,O-H的键长缩短,硝基的N-O键也由1.231和1.232 Å缩短至1.231和1.230 Å;对于keto,enol-1的O-H(1.006 Å)上的氢转移至氮上,形成N-H (1.029 Å),分子内氢键由O-H···N变为N-H···O,伴随着C=O的键长从1.332 Å 缩短至1.264 Å,且硝基的N-O键也由1.231和1.232 Å拉长至1.240和1.239 Å。从能量上来看,enol-1能量最低,enol-2,enol-3和enol-4的能量分别比enol-1高出6.1,7.4和10.4 kcal/mol,enol-4的能量高出10.4 kcal/mol,而keto的能量只比enol-1高出0.6 kcal/mol。如图3所示,从enol-1→enol-2的反应势能面曲线中也可以看出,enol-1→enol-2的扭转势垒为11.2 kcal/mol,而enol-2沿着苯并咪唑环与苯酚环的二面角扭转至enol-1也需要5.1 kcal/mol的能量。

Figure 2. Possible configurations of HBI-pNO2 optimized at the B3LYP/6-311 + G(d, p) level (bond length/Å and relative zero-point correction energy/kcal/mol)
图2. 在B3LYP/6-311 + G(d, p)水平下优化的HBI-pNO2的可能构型(键长/Å和相对零点校正能量/kcal/mol)

Figure 3. Isomerization potential energy surface curves of enol-1 and enol-2 calculated at B3LYP/6-311 + G(d, p) level (dihedral angle: C14-C12-C7-N20)
图3. 在B3LYP/6-311 + G(d, p)水平上计算的enol-1和enol-2异构的势能面曲线图
为进一步归属非极性溶剂中的吸收光谱,我们在B3LYP-TD/6-311 + G(d, p)水平上计算了enol-1的最大吸收波长、能量及振子强度(f),如表1所示。从表1可以看出:C6H6中,enol-1在S1态的最大吸收带位于384.3 nm,其振子强度为0.1014,对应于HOMO→LUMO的π→π*跃迁;S2态时,最大吸收波长在339.6 nm,振子强度为0.0031,属于π→π*跃迁(HOMO-1→LUMO);S4态时,最大吸收波长在316.6 nm,振子强度为0.7494,属于π→π*跃迁(HOMO→LUMO + 1);enol-1在S5态的最大吸收波长在313.5 nm,振子强度为0.2755,属于π→π*跃迁(HOMO-2→LUMO)。结合非极性溶剂中的紫外吸收光谱,发现其在S1,S4,S5态的理论计算值(384.3 nm, 316.6 nm, 313.5 nm)与实验值(332.0 nm, 319.2 nm, 291.2 nm)较吻合,因此在非极性溶剂苯(C6H6)中,enol-1存在于基态。图4是在B3LYP/6-311 + G(d, p)水平上计算的HBI-pNO2的基态势能面曲线,从图中可以看出,基态时,enol-1的能量比keto低了0.6 kcal/mol,且enol-1需克服4.1 kcal/mol的势垒质子转移形成keto。因此,我们认为基态时enol-1可以基态质子转移形成少量的keto。而在极性较大的溶剂中,我们猜测390~415 nm的吸收带可能是极性诱导的离子的吸收。

Table 1. Experimental and calculated singlet electronic transition energies, the corresponding orbital character and oscillator strengths of the enol-1 at B3LYP (TD)/6-311 + G(d, p) level (PCM=C6H6)
表1. 在B3LYP-TD (singlet, nstates = 15)/6-311 + G(d,p) (PCM, C6H6)水平下计算的enol-1的最大吸收波长(nm)、跃迁轨道及类型、能量(nm)和振子强度(f)
3.2. 不同pH中的紫外吸收光谱
为进一步确定HBI-pNO2在极性溶剂中390~415 nm处的吸收带是否为离子的吸收,我们通过实验分别获得了HBI-pNO2在酸性和碱性环境中的紫外吸收光谱如图5所示。从图5(a)中可以看出:甲醇中,HBI-pNO2的最大吸收波长分别在291.0、314.8、329.0及390.6 nm处。随着三氟乙酸(TFA)的加入,其吸收光谱发生改变,390.6 nm处的吸收带逐渐消失,形成了对应的阳离子;图5(b)中随着四丁基氢氧化铵([(Bu)4N]+OH−)的加入,形成对应的阴离子,伴随着390.6 nm处的吸收带愈加明显。由于阴离子的最大吸收波长与甲醇中的最大吸收波长相一致。由此我们得出结论:HBI-pNO2在极性溶剂甲醇中,390.6 nm处的吸收带对应阴离子的吸收。
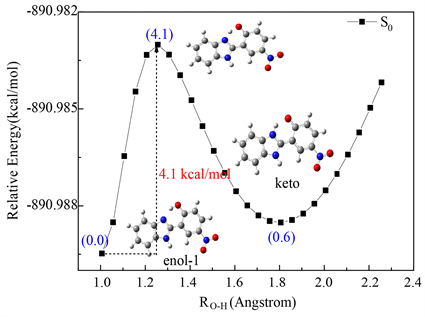
Figure 4. Potential energy curves of S0state for HBI-pNO2
图4. B3LYP/6-311 + G(d, p)水平上计算的HBI-pNO2的基态势能面曲线

Figure 5. Ultraviolet spectra for HBI-pNO2 in CH3OH with different volume of (a) TFA (b) [(Bu)4N]+OH−
图5. HBI-pNO2 (2.5 × 10−5 M)在CH3OH中随着(a)TFA的加入(b)[(Bu)4N]+OH−的加入而变化的稳态吸收光谱
为了指认HBI-pNO2在极性溶剂甲醇中的基态结构,对可能的阴离子进行了几何构型优化。图6是在B3LYP/6-311 + G(d, p)水平上计算可能的阴离子的键长和生成能,分别为羟基上的活泼氢与OH−结合形成的一价阴离子(DA1)、亚胺上的活泼氢与OH-结合形成的一价阴离子(DA2)、羟基上的活泼氢与OH−结合后扭转形成的一价阴离子(DA3)及两个活泼氢同时与OH-结合形成的二价阴离子(DA)。从优化的构型参数中发现:DA2与DA1相比,C-O键的键长从1.254 Å拉长至1.333 Å,硝基的键长由1.250 Å缩短至1.235 Å和1.236 Å,且DA2比DA1更平面;DA3与DA1相比,C-O键的键长从1.254 Å拉长至1.267 Å,硝基的键长由1.250 Å缩短至1.246 Å,且DA3更平面;对比DA1和DA,发现脱去两个质子的DA的苯并咪唑环与苯酚环扭转角度更大。从计算的能量可以看出,DA3及DA的生成能相对较低。
为进一步归属甲醇中的吸收光谱,我们在B3LYP-TD/6-311 + G(d, p)水平上计算了生成能较低的DA3及DA的最大吸收波长、能量、跃迁轨道及振子强度(f),如表2所示。从表2中可以看出:CH3OH中,DA3在S1态的最大吸收带位于435.3 nm处,其振子强度为0.2565,对应于HOMO-1→LUMO和

Figure 6. The optimized bond lengths in Å and relative energies (kcal/mol) for possible anion and binding energies (ΔH, kcal/mol) with PCM (solvent=CH3OH) model in S0 state at the B3LYP/6-311 + G(d, p)level level
图6. 在B3LYP/6-311 + G(d, p)水平下优化的阴离子的可能构型(键长/Å和生成能/kcal/mol)
Table 2. Experimental and calculated singlet electronic transition energies, the corresponding orbital character and oscillator strengths of DA3 and DA at B3LYP(TD)/6-311 + G(d, p) level (PCM=CH3OH)
表2. 在B3LYP-TD (singlet, nstates = 15)/6-311 + G(d, p) (PCM, CH3OH)水平下计算的DA3及DA的最大吸收波长(nm)、跃迁轨道及类型、能量(nm)和振子强度(f)
HOMO→LUMO的ππ*跃迁;S3态时,最大吸收波长在358.6 nm,振子强度为0.2634,属于ππ*跃迁(HOMO-2→LUMO和HOMO-1→LUMO)。与实验值(399.4 nm, 349.6 nm)相对较吻合,因此在极性溶剂中,HBI-pNO2除enol-1和keto之外,还存在少量的阴离子DA3。此外,从混合溶剂THF/H2O的紫外吸收光谱如图7所示,也可以看出,随着水的溶剂比的增大,其吸收光谱的变化规律与碱性环境的光谱变化一致,由此我们得出结论:在极性较大的溶剂中,极性诱导其阴离子DA3的生成,对应于390~420 nm的吸收。

Figure 7. Absorption spectra of HBI-pNO2 in THF/H2O mixtures with different fw values
图7. 在THF/H2O中不同溶剂比的紫外吸收光谱
4. 结论
通过2-(2'-羟基-5'-硝基)苯基苯并咪唑(HBI-pNO2)在不同溶剂中的稳态吸收光谱,结合密度泛函理论及含时密度泛函理论计算确定了,非极性溶剂中,HBI-pNO2以顺式醇构型存在于基态。借助HBI-pNO2在碱性环境中的稳态吸收光谱确定了,在极性溶剂中,HBI-pNO2在长波长处的吸收带对应于阴离子的吸收。实验结合理论计算指认了其阴离子结构为羟基脱氢以后扭转的一价阴离子结构DA3。
NOTES
*通讯作者。