1. 引言
环境污染和能源短缺是目前人类发展亟待解决的问题,新型能源的开发与利用成为当今的时代主题。光能、风能等可再生能源的转化与利用备受关注。其中,利用光能的光催化技术引起大量科研人员的研究。自从1972年,Fujishima和Honda [1] 第一次通过TiO2光解水产生氢气和氧气,光催化技术就大受科研人员关注。光催化技术通常被认为是一种可再生、安全、清洁的技术,其可通过催化分解水制备清洁的氢能源 [2] ,也可催化转化CO2制备合成气(其主要成分为CO和H2) [3] ,还可用于治理水体污染 [4] 等,由于应用广泛,因此半导体光催化剂得到深入研究。
近些年来,ZnIn2S4通过其自身在光催化领域中优异的物理化学性质,深受科研人员的青睐。ZnIn2S4是一种三元硫化物,具有层状结构、窄带间隙的半导体,带隙可调节(2.06 eV~2.85 eV),在可见光响应区域达570 nm [5] 。同时,与传统的金属硫化物如CdS和Sb2S3 [6] [7] 相比,ZnIn2S4的毒性小,且具有良好的化学稳定性、制备方法简单、来源丰富等优点,但是通过不断研究发现,ZnIn2S4仍存在一些缺陷,半导体ZnIn2S4的载流子分离效率低导致光催化效率低,且缺乏大量活性位点也限制了ZnIn2S4的光催化能力 [8] [9] [10] 。因此,广大科研人员对ZnIn2S4的修饰改性做出了许多科研研究,取得了大量的成果。科研人员发现,对可见光响应的半导体ZnIn2S4进行形貌调控 [11] [12] 、离子掺杂 [13] [14] 、贵金属沉积 [15] [16] 、半导体复合 [17] [18] 、缺陷工程 [19] [20] 等修饰改性可有效提高光吸收率,限制光生载流子复合,大大提高了其光催化性能。
2. ZnIn2S4的晶体结构及光催化作用机理
2.1. ZnIn2S4的晶体结构

Figure 1. Three crystal structures of ZnIn2S4: hexagonal (a), cubic (b), and rhombic (c) [21]
图1. ZnIn2S4的三种晶形结构:六方晶格(a),立方晶格(b),菱形晶格(c) [21]
ZnIn2S4是AB2X4族的三元硫化合物,具有理想的能带位置、独特的层状结构、良好的化学稳定性,是优异的半导体光催化材料。层状结构的ZnIn2S4具有三种晶体结构,如图1所示,分别为六方晶格、立方晶格、菱形晶格。相较于另两种相,六方相ZnIn2S4具有更理想的带隙结构,因此在光催化领域中更受关注。在ZnIn2S4的六方相中,原子沿c轴呈层状排列,Zn原子以四面体的方式与S原子结合,In原子以四面体或八面体的方式与S原子结合。这种原子排列提高了ZnIn2S4的光催化性能 [21] [22] 。
2.2. 半导体光催化作用机理
如图2所示,当半导体材料吸收合适的光子能量时,价带上的电子被激发到导带上,价带上形成对应的空穴,即产生电子–空穴对 [23] 。

Figure 2. Schematic diagram of energy band structure of semiconductor [23]
图2. 半导体的能带结构示意图 [23]
如图3所示为光催化降解机理示意图 [24] ,光生电子和空穴通过扩散作用由内部迁移到催化剂表面,很大一部分光生电子和空穴在迁移过程在内部或表面发生复合,以热能或其他形式的能量释放,另一方面,环境中有合适的捕获剂或表面缺陷时,光生电子和空穴会移动到催化剂表面,由于光生电子具有强还原能力,会在材料表面被O2所捕获形成超氧自由基∙O2−;空穴(h+)具有强氧化能力,会与半导体表面上的H2O和OH−发生反应,生成∙OH。产生的这些活性自由基(h+、∙O2−、∙OH)具有强氧化性,从而发生氧化还原反应。在光催化反应中,催化制氢、降解污染物、催化转化二氧化碳的原理基本类似,区别于最终氧化还原反应。
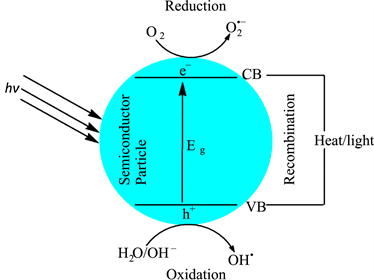
Figure 3. Mechanism of semiconductor photocatalytic reaction [24]
图3. 半导体光催化反应机理 [24]
3. ZnIn2S4及其改性材料在光催化领域中的发展
由于ZnIn2S4具有较窄的禁带宽度、独特的层状结构、良好的稳定性、无毒无污染,且易于制备来源丰富,有利于得到推广使用,因此在光催化领域中大受研究者们关注。但该材料的光催化效率并不高,限制了其大量使用,其主要原因是光生载流子易于复合,缺乏大量活性位点。因此,需要对其进行修饰改性,提高光催化性能。下面将通过形貌调控、元素掺杂、贵金属沉积、半导体复合,缺陷工程五个方面介绍ZnIn2S4的修饰改性对光催化性能的影响。
3.1. 形貌调控
形貌调控是一种通过改变合成材料的方法,在其适宜条件下得到不同形状尺寸的材料来改善其光催化性能的方法。自2003年,Lei等人 [5] 第一次通过水热法合成ZnIn2S4纳米球以来,关于ZnIn2S4形貌调控的研究一直是科学家们研究的热点。形貌调控一方面可通过调节ZnIn2S4的比表面积和吸光度,提高对光的吸收效率;另一方面,通过增加暴露的表面活性位点,从而提高光催化活性。目前,通过水热法、溶剂热法、微波辅助法等已经合成得到多种不同形貌的ZnIn2S4,如纳米微球、微簇 [25] 、纳米片 [26] 、纳米带、纳米管 [27] 等。
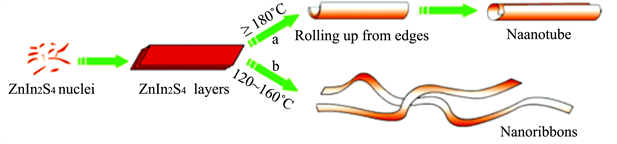
Figure 4. Schematic diagram of growth mechanism for ZnIn2S4 nanotubes (a) and nanoribbons (b) [28]
图4. ZnIn2S4纳米管(a)和纳米带(b)的生长机制示意图 [28]

Figure 5. TEM images of the ZnIn2S4 samples obtained at 180˚C after solvothermal reaction for 2 h (a), 8 h (b), and 12 h (c), respectively, confirming the rolling growth process of ZnIn2S4 nanotubes from lamellar structures [28]
图5. 溶剂热反应2 h (a)、8 h (b)和12 h (c)后在180℃下获得的ZnIn2S4样品的TEM照片,证实了ZnIn2S4纳米管从层状结构的滚动生长过程 [28]
Gou等人 [28] 通过以吡啶为溶剂的溶剂热法合成了一维三元硫化合物ZnIn2S4纳米结构。实验结果表明,一维ZnIn2S4纳米结构包括成核和自生长两步骤,生成的ZnIn2S4层状中间体可以通过在不同的条件下形成管状或纳米带(如图4所示)。通过受控实验和TEM观察结果,我们发现温度和持续时间热处理对最终形态至关重要。当反应温在160℃和120℃,仅能形成纳米带,在180℃的高温下,溶剂热处理足够的持续时间后可形成ZnIn2S4纳米管,如图5所示。上述结果表明,ZnIn2S4纳米管和纳米带可以通过控制选择性合成溶剂热反应的温度和持续时间。此外,制备的ZnIn2S4纳米管和纳米带材料在光催化反应中带来了良好的反应速率,形貌调控不仅调节了ZnIn2S4的吸光度,提高了对光的吸收效率,而且纳米管和纳米带更有效地增加了表面活性位点提高反应速率,从而提高光催化活性。紫外–可见吸收光谱也表明,在从紫外光到可见光的宽范围内具有很强的吸收能力,并且它们的带隙与尺寸和形貌有一定关系,从而大大提高了光催化性能。由于形貌调控可与其他改性方法共同使用,使得这种纳米管、纳米带材料的研究更加广泛。
Shen等人 [29] 通过水热法合成的ZnIn2S4 (ZIS)样品在不同水热处理时间下,其形貌各不相同。水热处理1 h时,ZnIn2S4微晶形成由许多ZnIn2S4花瓣组成的微球;6 h~12 h时,ZnIn2S4表现为直径为1至2 μm的均匀微球,也包括无数的花瓣;24时,得到的ZnIn2S4产物主要表现为微团簇。48 h时,得到的ZnIn2S4产物表现为不同形态的混合物,包括微团簇和微花。产物ZnIn2S4的形貌随水热处理的时间变化而变化,然而,ZnIn2S4产品的这种形态差异的原因和详细机制尚不清楚。但水热处理的时间对ZnIn2S4产品的结构和形貌有着重要影响,从而影响其光催化性能。ZIS-1.8-y (y = 1 h~48 h)光催化剂上的光催化制氢速率如图6所示。在15小时的光催化反应过程中,随着水热处理时间的延长,ZIS-1.8-y (y = 1 h~48 h)光催化剂的光催化活性降低。表明ZIS-1.8-y (y = 1 h~48 h)光催化剂的不同形态可能是影响光催化性能的一个因素。与呈现微团簇形态的ZIS-1.8-y (y = 24 h~48 h)光催化剂相比,具有微球状形态的ZIS-1.8-y (y = 1 h~12 h)光催化剂增加了表面活性位点,从而具有更高的光催化活性。也就是说,微球形态在一定程度上有利于提高ZnIn2S4光催化剂的光催化活性。由于某些材料在其他改性方法中不能保持一维、二维的形态,因此这种形貌调控的结果对三维材料中光催化反应速率的提高来说非常重要。
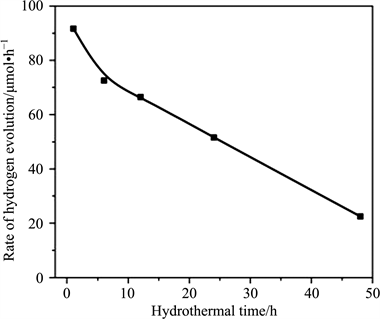
Figure 6. Rates of hydrogen evolution over ZnIn2S4 photocatalysts synthesized by hydrothermal method for different times [29]
图6. 不同时间水热法合成的ZnIn2S4光催化剂的析氢速率 [29]
3.2. 元素掺杂
元素掺杂是光催化剂中较简单的一种改性方法,将杂质元素引入到半导体晶格内部形成新电荷,增加缺陷或者产生新的杂质能级来改善光催化性能。一方面,通过改变能带结构可缩小带隙,拓宽光吸收区域;另一方面,掺杂会在催化剂表面形成缺陷,俘获电子,且抑制光生载流子的复合,大大提高光催化性能。
Shen等人 [30] 通过简易的水热法在CTAB辅助下合成了一系列不同量Cu2+掺杂的ZnIn2S4光催化剂,铜的重量含量从0 wt%到2.0 wt%不等。并通过实验详细讨论了铜掺杂对催化剂ZnIn2S4形貌、光学性质、光催化活性的影响。Cu2+的加入直接影响了催化剂Cu-ZnIn2S4的形貌,随着铜掺杂量的不断增多,ZnIn2S4微球的直径不断减小,接着微球被部分破坏,甚至彻底破坏;即六边形的层状结构ZnIn2S4,以及微球的形状,都逐渐被增加的Cu2+掺杂剂破坏。且随着Cu2+量的不断增加,ZnIn2S4光催化剂的析氢速率也在逐渐增大,经过分析其原因为,Cu(X)-ZnIn2S4 (X为Cu的重量分数)的可见光吸收边缘波长不断得到增加,拓宽了可见光吸收范围,相对应的带隙不断缩小。导致了光吸收效率增大,因此总体增加了光析氢速率,因此得到,利用一定的合成方法对ZnIn2S4进行元素掺杂可改变能带结构缩小带隙、拓宽光吸收区域,这是一种非常有效的改性手段。如图7所示为在可见光照射下,Cu(X)-ZnIn2S4光催化剂的析氢速率。未掺杂Cu2+的ZnIn2S4上的析氢速率为26.1 μmol·h−1,当0.5 wt%的Cu2+掺杂ZnIn2S4时,其获得最高光催化活性,析氢速率达到151.5 µmol·h−1。这也为研究人员通过其他的合成手段进行元素掺杂时,使ZnIn2S4获得良好的光吸收范围提供了一种思路。
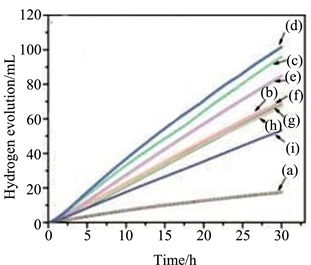
Figure 7. Photocatalytic H2 evolution under visible light-irradiation over Cu (X)-ZnIn2S4; the values of X were (a) 0.0 wt%, (b) 0.1 wt%, (c) 0.3 wt%, (d) 0.15 wt%, (e) 0.17 wt%, (f) 0.9 wt%, (g) 1.2 wt%, (h) 1.6 wt%, (i) 2.0 wt% [30]
图7. Cu(X)-ZnIn2S4可见光下光催化析氢;X值为(a) 0.0 wt%,(b) 0.1 wt%,(c) 0.3 wt%,(d) 0.15 wt%,(e) 0.17 wt%,(f) 0.9 wt%,(g) 1.2 wt%,(h) 1.6 wt%,(i) 2.0 wt% [30]

Figure 8. Schematic diagram of the band structure of the pristine ZnIn2S4 and N-doped ZnIn2S4 samples [31]
图8. 原始ZnIn2S4和N掺杂ZnIn2S4样品的能带结构示意图 [31]
Du等人 [31] 通过将0.4 mmol Zn (CH3COO)2·2H2O, 0.8 mmol InCl3和1.6 mmol硫代乙酰胺溶解在30 ml的含有不同剂量N,N-二甲基甲酰胺(DMF)的水溶液中,采用溶剂热法合成了一系列样品。根据DMF的不同剂量(0 ml、1 ml、6 ml、12 ml、15 ml和18 ml),相应的样品分别表示为ZnIn2S4 (ZIS)、NZIS-1、NZIS-2、NZIS-3、NZIS-4和NZIS-5。对这些样品的光催化活性进行了一系列实验验证,并进行讨论。在可见光照射下分析其光催化产氢性能,发现NZIS-3的活性最高,产氢速率高达11,086 μmol·g−1·h−1,是ZIS的13.8倍(801 μmol·g−1·h−1)。光催化活性得到显著提高的原因是(如图8所示):N掺杂在ZIS后不仅增加了VB的宽度,且N掺杂的缺陷状态促进了空穴的迁移率,从而有效地分离了电子空穴对,延长了光生载流子的寿命;而且还提高了CB的位置,为光催化反应提供了更多的光生电子,且加速了光生电子的迁移。这些结果均得到了理论计算的支持。由此,我们可采用不同的合成方法,以及在不同的合成条件下控制不同的元素对ZnIn2S4进行掺杂,从而有效地促进光催化反应中电子空穴对的分离,以及加快空穴的迁移,从而使电子空穴对的利用率提高,大大加快光催化反应的效率。
3.3. 贵金属沉积
通常情况,当光催化剂ZnIn2S4吸收光能激发产生电子空穴对时,大量光生电子空穴对并不能迁移到催化剂表面发生氧化还原反应,而是迅速复合以热能的形式释放。而引入贵金属可进行有效改善,贵金属如Ag、Pt、Au等由于其费米能级一般低于半导体ZnIn2S4的费米能级,光生电子会优先转移到金属中,从而抑制光生载流子的复合,贵金属还由于表面等离子体共振效应(SPR)增强半导体对可见光的吸收能力,增大了ZnIn2S4对可见光的吸收效率。
An等人 [32] 制备Au@Pt/ZnIn2S4 (ZIS)的过程分为三个步骤,首先,通过柠檬酸盐还原法合成Au16 (NPs),然后通过种子生长方法制备Au16@Pt (NPs),合成的Au16@Pt (NPs)具有带负电荷的表面;其次,通过溶剂热法合成ZIS,并在其表面覆盖一层带有正电荷的APTES进行修饰,最后,APTES修饰的ZIS与Au16@Pt (NPs)混合获得Au16@Pt/ZIS催化剂(如图9所示)。通过光催化制氢实验发现,在10小时的光照下,Au16@Pt/ZIS的产氢速率和产量分别为4174.7 μmol·g−1 h−1,41,747 μmol·g−1,约为纯ZIS的十倍,Au46@Pt/ZIS的产氢速率也达到了2094 μmol·g−1 h−1,约为纯ZIS的五倍。其结果表明,通过在ZIS上负载Au@Pt组件,可显著提高其光催化性能。通过分析Au@Pt/ZIS的光催化制氢机制(如图10所示)可得出结论,Au的SPR激发和ZIS的带隙激发在可见光照射下同时发生,导致光生载流子增加;同时,Pt的引入可以有效地从Au (NPs)和ZIS中分离和积累电子,不仅可促进光生载流子复合,还可作为H+还原位点,从而进一步增加H2的产量。该研究表明,由具有等离子体和催化作用的双金属纳米粒子组成半导体上的组件将是一种提高光催化性能的有前途且有效的方法。
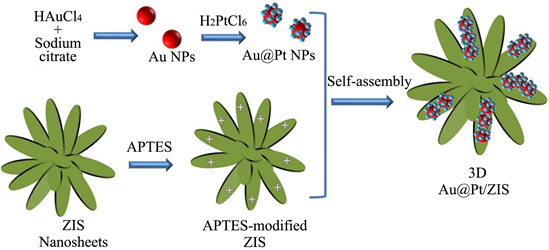
Figure 9. Schematic diagram of the formation process of Au@Pt/ZIS [32]
图9. Au@Pt/ZIS形成过程示意图 [32]
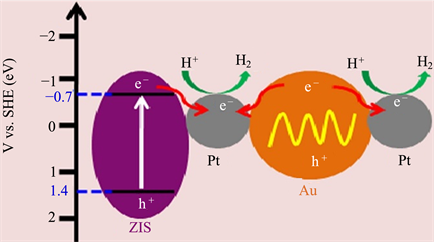
Figure 10. Reaction mechanism for photocatalytic H2 production in Au@Pt/ZIS system [32]
图10. Au@Pt/ZIS体系中光催化制氢的反应机理 [32]
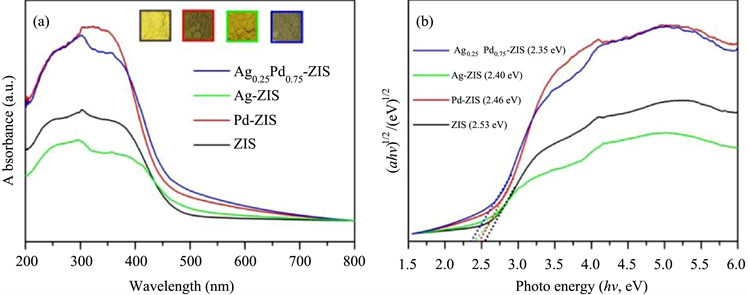
Figure 11. (a) UV-Vis DRS (Inset is the color images of as-prepared samples); (b) Relationship plots of transformed Kubelka-Munk function versus the light energy for pure ZIS, Ag-ZIS, Pd-ZIS and Ag0.25Pd0.75-ZIS [33]
图11. (a) UV-Vis DRS (插图是所制备样品的彩色图像);(b) 转换后的Kubelka-Munk函数与纯ZIS、Ag-ZIS、Pd-ZIS和Ag0.25Pd0.75-ZIS的光能的关系图 [33]
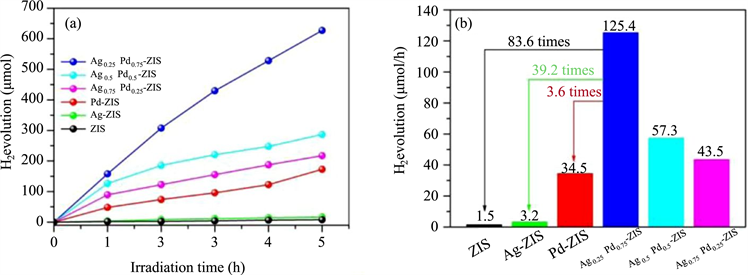
Figure 12. (a) Changing tendency of H2 evolution amount with time and (b) H2 evolution rate of ZIS and AgxPd1−x ZIS (x = 0, 0.25, 0.5, 0.75 and 1.0) samples under visible light (λ > 420 nm) [33]
图12. ZIS和AgxPd1−x-ZIS (x = 0, 0.25, 0.5, 0.75和1.0)样品在可见光(λ > 420 nm)下,(a) H2析出量随时间变化趋势及(b) H2析出速率 [33]
Liu等人 [33] 通过化学还原法在ZnIn2S4表面负载贵金属Ag-Pd合金制备了AgxPd1−x/ZnIn2S4。通过对纯ZnIn2S4、Ag-ZnIn2S4、Pd-ZnIn2S4以及Ag0.25Pd0.75-ZnIn2S4的紫外可见漫反射光谱进行研究发现,如图11所示,贵金属Ag、Pd和Ag-Pd合金均明显提高了ZnIn2S4的可见光吸收能力。在光催化制氢实验中,在ZnIn2S4表面上形成的Ag-Pd合金由于双金属协同效应而显著增强其产氢效率。如图12所示,Ag0.25Pd0.75-ZIS样品产氢速率达到125.4 μmol/h,分别比ZIS、Ag-ZIS和Pd-ZIS高83.6倍、39.2倍和3.6倍。科研人员通过一系列实验验证,发现Ag-Pd合金提高光催化制氢的原因为:在可见光照射下,ZnIn2S4被激发产生光生载流子。由于Ag-Pd合金的费米能级比ZnIn2S4的CB电位更低,ZnIn2S4中的光生电子会通过肖特基势垒迁移到Ag-Pd中,从而促进了光生载流子的分离,延长了光生载流子的寿命;同时,Ag-Pd合金也提高了其可见光吸收能力,进而显著提高了光催化制氢的效率。
Shi等人 [34] 通过采用光化学途径合成具有Pt单位点的2D的ZnIn2S4 (h-ZIS),由于光诱导反应比传统退火方法更温和可控,因此h-ZIS的结构可以很好地保留,且不会产生空位缺陷。此种方法合成的负载Pt的h-ZIS制氢效率最高可达纯h-ZIS的17.8倍,大大提高了其光催化性能。经探讨,制氢速率的提高是由于原子突起状Pt原子在光催化中具有三重作用。1) 单个Pt原子调整了h-ZIS的能带结构,提供了更多的光生载流子;2) 由于分散的Pt原子的费米能级低于ZnIn2S4的CB电位,可充当电子陷阱捕获光生电子,促进了光生载流子的分离和迁移;3) Pt原子降低了H2析出过电位,加速了催化反应的速率。由于只需使用极少量的Pt,因此该研究为贵金属沉积降低成本提供了很好的方法,使其可能得到推广使用。
3.4. 构建异质结
单一的ZnIn2S4半导体由于光生载流子难分离,易复合,利用率低限制了其光催化性能,利用构建异质结的方法可有效改善此问题。此方法通过利用两种半导体能级结构差异,使得光生载流子分别迁移至各自的活性位点,大大加快了光生载流子迁移的速率,抑制了光生载流子复合,提高了其光催化性能。

Figure 13. (a) The representative fabrication process of ZIS/HCNT heterostructures; SEM images of HCNT (b), ZIS (c) and ZIS/HCNT-2 (d); TEM image (e), magnified TEM image (f) and HRTEM image (g) of ZIS/HCNT-2; (h) HAADF image and the corresponding elemental mapping images of C, N, Zn, In and S for the ZIS/HCNT-2 [35]
图13. (a) ZIS/HCNT异质结构的合成制备过程;HCNT (b)、ZIS (c)和ZIS/HCNT-2 (d)的SEM图像;ZIS/HCNT-2的TEM图像(e)、放大的TEM图像(f)和HRTEM图像(g);(h) ZIS/HCNT-2的HAADF图像和相应的C、N、Zn、In和S元素映射图像 [35]
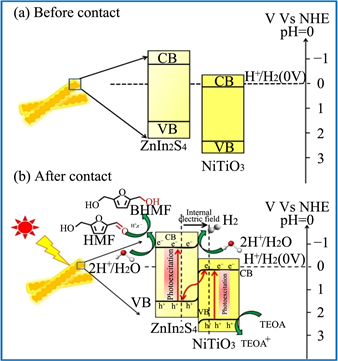
Figure 14. Pictorial representation of (a) before and (b) after heterojunction formation for Z-scheme mechanism for photocatalytic H2 generation and subsequent HMF reduction by NiTiO3/ZnIn2S4 hierarchical nanostructures [36]
图14. 异质结形成之前(a)和(b)之后的图形表示,用于光催化H2生成和随后通过NiTiO3/ZnIn2S4分级纳米结构还原HMF的Z方案机制 [36]
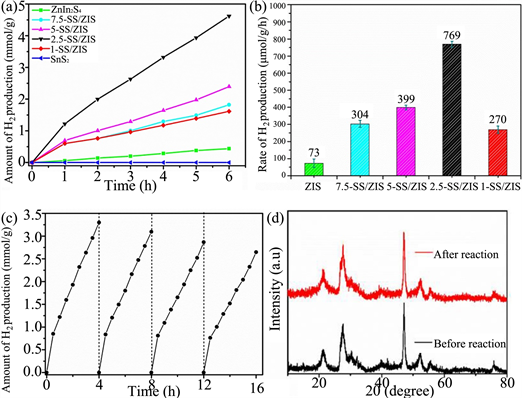
Figure 15. (a) H2 production amount and (b) H2 production rate within 6 h of all the samples; Error bars represent the standard deviation for three parallel experiments; (c) Cyclic H2-production over 2.5-SS/ZIS photocatalyst; (d) XRD patterns of 2.5-SS/ZIS before and after irradiation [38]
图15. 所有样品6 h内(a) H2产生量和 (b) H2产率;误差线代表三个平行实验的标准偏差;(c) 2.5-SS/ZIS光催化剂上的循环产氢,(d) 辐照前后2.5-SS/ZIS的XRD图谱 [38]
Li等人 [35] 通过原位生长法制备了基于g-C3N4纳米管的S型异质结(ZIS/HCNT)催化CO2还原。ZnIn2S4纳米片在g-C3N4纳米管表面原位生长,导致ZnIn2S4和g-C3N4之间紧密作用,其形成的核壳结构可同时拥有一维特征的形态优势和S型异质结电荷扩散的长处。ZIS/HCNT S型异质结的代表制造过程如图13(a)所示,其通过煅烧,水热处理,热缩聚一系列过程合成。图13(b)~(c)所示,HCNT具有由超薄g-C3N4纳米片(约100 nm)组装而成的均匀六角形微管结构,ZIS倾向于聚集成直径约2 μm的花状微球结构,由许多交错的薄片组成。从图13(d)可以看出,超薄ZIS纳米片在HCNT管表面均匀而致密地生长,形成核壳结构。图13(e)~(f)揭示了ZIS/HCNT的核壳结构以及ZIS和HCNT之间的紧密界面。样品ZIS/HCNT独特的分层结构具有更高的比表面积,为光催化反应提供了许多活性位点。核壳对齐的接触可以大大缩短光生载流子的垂直扩散距离,从而抑制电荷复合,独特的S型异质结大大促进光生载流子的迁移和分离。实验数据显示,ZIS/HCNT表现出显著提高的CO2到CO的光催化转化率(883 μmol·g−1·h−1),分别是HCNT的13倍和ZIS的2.4倍。
Dhingre等人 [36] 通过原位沉积的方法合成制备了NiTiO3/ZnIn2S4异质结,即通过在NiTiO3微棒上原位沉积ZnIn2S4纳米片制备。并通过一系列光催化制氢,光催化降低HMF实验探究了NiTiO3/ZnIn2S4异质结的催化机理,如图14所示。进一步分析构成的Z型异质结提高光催化性能的本质,发现异质结内部电场、能带弯曲和库仑排斥等各种因素会阻止光生载流子复合,有利于光生载流子的分离,从而提高ZnIn2S4的催化活性。NiTiO3/ZnIn2S4异质结光催化剂在高效制氢和还原衍生物HMF成高价值化学品BHMF均表现出优异的光催化活性,突出了Z型异质结在两半导体接口处的促进光生载流子分离和迁移中的作用。
研究者邵博宇等人 [37] 通过简单的溶剂热法在立方体In2O3六个表面上生长出不同形貌的ZnIn2S4,成功构建了3D/2D的In2O3/ZnIn2S4异质结。在光催化还原CO2的一系列测试中,其表现出优异的光催化活性,CO产率高达2064.5 μmol·g−1 h−1,超过单一的ZnIn2S4和In2O3,即使是3D/0D的In2O3/ZnIn2S4的异质结,其CO产率也仅为1231.7 μmol·g−1 h−1。一方面,不仅提供了高速的异质结通道促进光生载流子的分离与迁移,提高了光生载流子的利用效率。另一方面,其独特的三维/二维结构提供了更多的光接触面积,增加了可见光的吸收;还有更大的界面接触面积,使得大量活性位点暴露在催化剂上。为不同形貌纳米复合结构型异质结的构建提供思路。

Figure 16. (a) PL spectra, (b) Photocurrent responses and (c) EIS Nyquist plots of ZnIn2S4, SnS2, 2.5-SS/ZIS [38]
图16. (a) PL光谱,(b)光电流响应和(c) ZnIn2S4、SnS2、2.5-SS/ZIS的EIS Nyquist图谱 [38]
Geng等人 [38] 通过水热法合成了具有Type 2型异质结的SnS2/ZnIn2S4光催化剂,并通过改变SnS2 (SS)与ZnIn2S4 (ZIS)的质量比,合成了一系列SnS2/ZnIn2S4,其质量比分别为1%、2.5%、5%和7.5%时,记录为1-SS/ZIS、2.5-SS/ZIS、5-SS/ZIS和7.5-SS/ZIS。在可见光照射下,对上述材料以及纯ZIS进行了光催化制氢性能的测试,如图15所示,产氢速率的最高的为2.5-SS/ZIS,达到769 μmol·g−1 h−1,约为纯ZIS的10.5倍(产氢速率为73 μmol·g−1 h−1)。且通过四轮循环实验评估了其稳定性,产氢速率仅略微下降;其衍射峰相同也表明其具有良好的稳定性。其光催化下显著提高的原因可能是由于SS/ZIS异质结的形成,使得在SnS2和ZnIn2S4界面处光生载流子迅速分离和转移,抑制了复合。通过对材料的测试得到证明,如图16(a)所示,2.5-SS/ZIS的发射峰远弱于SnS2和ZnIn2S4,证明了光生载流子在2.5-SS/ZIS中复合受到显著阻碍;如图16(b)所示,2.5-SS/ZIS表现出更强的光电流密度,表明2.5-SS/ZIS复合材料中激发的电子和空穴的分离效率提高;如图16(c)所示,EIS中2.5-SS/ZIS表现出最小的圆弧半径,意味着电荷2.5-SS/ZIS的转移阻力是最低的,即电子的转移速度得到显著增加。
3.5. 缺陷工程
近年来,在光催化剂制备过程中,通过调控制备方法、条件等,在光催化剂中引入空位被证明是一种可有效提高光催化活性的技术。空位的引入会在半导体中形成缺陷能级,有利于拓宽光吸收范围,产生更多的光生载流子,且空位可作为俘获光生电子的电子陷阱,促进光生载流子的分离与迁移,同时作为活性位点,改善了电荷密度和降低催化反应的能量势垒。
Wang等人 [39] 运用低温油浴法通过控制反应时间制备了有缺陷的ZnIn2S4纳米片,有S空缺(Vs)的ZnIn2S4 (ZIS)纳米片记为(Vs-ZIS),并证明了所制备的Vs-ZIS材料是光催化CO2还原成合成气(CO和H2)的理想催化剂。在光催化CO2还原合成气实验中,3 h内,原始ZIS气体产率仅为4.71 mmol·g−1,选择性也不理想(CO和H2比例为1:4.18);而Vs-ZIS合成气量达到22.27 mmol·g−1,比原始ZIS高出约4.73倍,CO/H2比约为1:1。通过详细讨论得知,构建的S空位形成缺陷能级,拓宽了光吸收区域,光吸收能力得到提升,其次,S空位还可作为捕获光生电子的电子陷阱,有效抑制光生载流子复合,大大加快光生电子空穴对的分离与迁移。同时,S空位的位置位于Zn原子周围。负离子空位会降低锌阳离子的平均氧化状态。低配位的锌和空位均增加CO2的吸附能力,导致了更高的CO选择性,为合成气选择性的制备提供了有力的证明。如图17所示。

Figure 17. Schematic illustration of reaction mechanism over Vs-ZIS (a) and pristine ZIS (b) [39]
图17. Vs-ZIS (a)和原始ZIS (b)的反应机理示意图 [39]
Laponite-RD (Lap)是一种层状硅酸盐粘土,Liu等人 [40] 通过以Laponite为模板(如图18所示),采用胶体水热法合成具有Zn缺陷的ZnIn2S4 (ZIS)-Lap异质结构纳米材料,由于锌(Zn2+)和镁(Mg2+)离子具有相似的离子半径,从Lap八面体层中浸出的Mg2+可以部分替代ZnIn2S4中的Zn2+,形成Zn缺陷。我们通过在可见光照射下甲基橙(MO)的降解被用来确定样品的光催化活性,发现ZIS-Lap-Mg2+样品的表观量子效率升高。经分析得知,用作电子俘获中心的ZnIn2S4层中引入Zn空位提高了电荷分离效率,电子空穴对传输能力增强。此外,添加游离Mg2+可减弱合成过程中的浸出过程,减少Mg2+的浸出,从而减少了Zn缺陷的形成,这已被证明可以提高电荷分离效率。这项工作提供了一种在二维和少层纳米材料中引入缺陷的新方法。
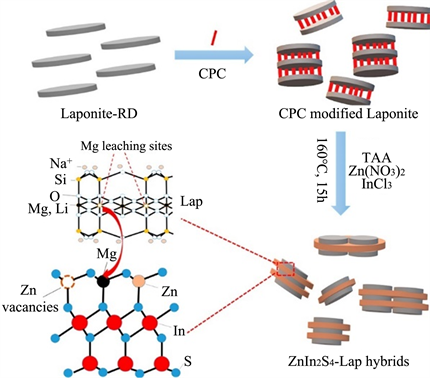
Figure 18. Synthesis of ZIS-Lap nanohybrids [40]
图18. ZIS-Lap纳米杂化物的合成 [40]
4. 总结与展望
本文从形貌调控、元素掺杂、贵金属沉积、半导体复合、缺陷工程五个方面较为详细地描述了光催化剂ZnIn2S4的修饰改性,分析了改性过后的ZnIn2S4的优缺点。针对光生载流子复合快、利用率低、催化反应慢等都做出了相应的改性措施,明显提高了ZnIn2S4的光催化性能,使得ZnIn2S4材料在光催化制氢、光催化降解污染物等大量应用。但是,目前ZnIn2S4的修饰改性材料仍存在不足:1) ZnIn2S4的掺杂改性仍以单一离子为主,且目前对ZnIn2S4离子掺杂的研究仍十分少有。2) 在ZnIn2S4的修饰改性中,可将形貌调控与其他几种方式结合提高其光催化性能,但其他几种方式如元素掺杂与构建异质结进行结合起到协同作用,改善ZnIn2S4性能的相关报道目前较少。3) 光催化降解污染物时,污染物种类较多,但ZnIn2S4的改性材料只能处理某种或某些污染物,功能较为单一,在ZnIn2S4光降解污染物的应用中仍需更多研究。4) 对ZnIn2S4缺陷工程的研究目前较少,且该改性方法具有十分广大的前景。
NOTES
*通讯作者。