1. 引言
拟南芥(Arabidopsis thaliana)是十字花科(Cruciferae)芥菜科的一种小型开花植物。它分布在世界各地,最早在16世纪由约翰内斯·塔尔(Johannes Thal)报道。五十多年来,它已被广泛用于研究植物突变和经典的遗传分析。拟南芥现在已成为植物生物学不同研究领域的模式生物。
拟南芥是一种具有2n = 10条染色体的二倍体植物。拟南芥成为第一个完成全基因组测序植物是基于以下几点优势:1) 基因组约为120 Mb,且结构简单,重复序列少;2) 从种子萌发到结实期为6周,世代时间短;3) 产生大量种子。测序工作由国际合作组织拟南芥基因组计划(Arabidopsis Genome Initiative, AGI)完成。拟南芥作为一种重要的农业作物来说,是一种无价的资源。
Cys2His2 (C2H2)型锌指蛋白是广泛存在于真核转录因子中的DNA结合模体。锌指结构是由两个或三个β折叠和一个α-螺旋组成的短蛋白质序列。位于特定位置的两个半胱氨酸和两个组氨酸残基与锌离子结合以稳定结构。另外四个氨基酸残基定位在α-螺旋的N端,通过与DNA主槽中的氢供体和受体相互作用参与DNA结合。一个蛋白质中锌指的数量可以在很大范围内变化,从而使目标DNA序列具有可变性。锌指蛋白除具有DNA结合作用外,还具有蛋白质与蛋白质、RNA与蛋白质的相互作用。在大多数情况下,含有C2H2型锌指蛋白的蛋白质是基因表达的转运调节因子,在细胞发育、分化和抑制恶性细胞转化(抑瘤)等过程中起着重要作用 [1]。
本实验通过对拟南芥IDZ基因家族进行生物信息学分析,揭示拟南芥IDZ的结构及进化特点,为后期功能基因挖掘及深入功能分析提供基础数据。
2. 材料与方法
2.1. 拟南芥IDZ基因家族的选取及理化分析
拟南芥IDZ基因家族信息来源于TAIR数据库(https://www.arabidopsis.org/index.jsp),并通过NCBI获取IDZ基因家族蛋白质序列信息,并通过ExPaSy [2] [3] [4] (https://web.expasy.org/compute_pi/)在线工具进行理化性质分析
2.2. 拟南芥IDZ基因家族的多序列比对
运用T-coffee [5] 在线多序列比对工具(http://tcoffee.crg.cat/apps/tcoffee)对拟南芥IDZ基因家族序列做多序列比对,将结果导入ESPript 3.0 [6] (https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi),Secondary structure depiction的Parameters参数设置为Sec.structure labels: α1,β2,α2,β2,…,Sequence similarities depiction parameters设置为%Equivalent,结果经过检验验证。
2.3. 拟南芥IDZ基因家族系统进化树构建
使用ClustalW对拟南芥IDZ基因家族序列进行多序列比对,参数设置为Pairwise Alignment的Gap Opening Penalty: 10,Gap Extension Penalty: 0.1;Multiple Alignment的Gap Opening Penalty: 10,Gap Extension Penalty: 0.2;Protein Weight Matrix设置为Identity,Residue-specific Penalties设置为ON,Hydrophilic Penalties设置为ON,Gap Separation Distance: 4,End Gap Separation 设置为off,Use Negative Matrix设置为OFF,Delay Divergent Cutoff(%):30。将结果用MEGA 7 [7] 构建系统进化树,构建采用临近算法(Neighbor-Joining, NJ),Bootstrap检验1000次,Model/Method设置为p-distance,Rates among Sites设置为Uniform rates, Gaps/Missing Data Treatment设置为Partial deletion,Site Coverage Cutoff(%):50,得出的系统进化树设置Hide valueslowerthan 60%。
2.4. 拟南芥IDZ基因家族的结构分析
使用MEME [8] (https://meme-suite.org/meme/tools/meme)在线工具对拟南芥IDZ基因家族进行分析,参数设置motif为10。将结果与进化树导入TBtools [9] 将蛋白质结构域可视化,使用Gene Structure View (Advanced)功能,勾选Fill in Gradient Mode和Motif Num,将Width设置为1500,Hight设置为300。
使用Pfam [10] (http://pfam.xfam.org/)数据库进行数据分析,可得蛋白结构域信息。
2.5. 拟南芥IDZ基因家族的染色体定位分析
从TAIR数据库上拉取拟南芥IDZ基因家族Genomic Locus相关信息制表,使用MG2C 2.0 [11] (http://mg2c.iask.in/mg2c_v2.0/)导入相关信息绘制染色体定位图。
2.6. 蛋白互相作用网络构建
将蛋白质名称输入STRING v11.5 [12] (https://string-db.org/)数据库分析可构建出蛋白互相作用网络,利用分析功能可得网络统计数据,以及GO基因富集等相关分析结果。
2.7. 蛋白三维结构分析
将蛋白质名称输入Uniprot [13] (http://beta.uniprot.org/)数据库可得蛋白质三维结构,根据AlphaFold [14] 产生的置信评分(plDDT)不同,进行注释分析。
3. 结果与分析
3.1. 拟南芥IDZ基因家族的选取及理化分析
从TAIR数据库获取拟南芥IDZ家族成员共11个 [15] [16],利用ExPaSy在线工具理化性质分析显示(表1),该蛋白家族分子量大小为64463.70 (AtIDZ2)至44513.05 (AtIDZ8),等电点为9.63 (AtIDZ3)至8.36 (AtIDZ5)。

Table 1. Physicochemical properties of IDZ gene family in Arabidopsis thaliana
表1. 拟南芥IDZ基因家族理化性质
3.2. 拟南芥IDZ基因家族的多序列比对
利用T-coffee在线多序列比对工具(http://tcoffee.crg.cat/apps/tcoffee)对拟南芥IDZ基因家族序列做多序列比对,将结果导入ESPript 3.0 (https://espript.ibcp.fr/ESPript/ESPript/)可得多序列比对可视图(图1)。由结果可知在序列40-200、350-370、410处存在高度保守区段。

Figure 1. Sequence alignment of IDZ gene family in Arabidopsis thaliana
图1. 拟南芥IDZ基因家族序列比对结果
3.3. 拟南芥IDZ基因家族的进化树分析
拟南芥IDZ基因家族系统进化树上都为旁系同源,是原始基因复制分离而产生的同源基因,由图(图2)可知原始的基因因复制产生了分支,随后又各自复制产生多分支,在进化过程中现在IDZ家族成员里AtIDZ4的基因与原始基因的差异最小,而AtIDZ2、AtIDZ11的基因与原始基因差异最大。

Figure 2. Phylogenetic tree of IDZ gene in Arabidopsis thaliana (NJ)
图2. 拟南芥IDZ基因的系统进化树(Neighbor-Joining Method, NJ)
3.4. 拟南芥IDZ基因家族的结构分析
根据TBtools结果图(图3)可知拟南芥IDZ基因家族都有motif1-7、motif10,motif9仅存在于AtIDZ9、AtIDZ10头部和AtIDZ2尾部,motif8仅存在于AtIDZ4、AtIDZ5尾部,因此这五个蛋白质也许有其特殊功能。
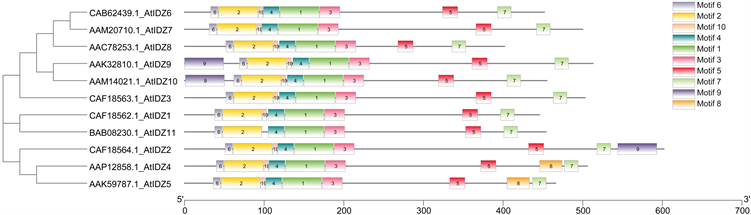
Figure 3. IDZ protein motif in Arabidopsis
图3. 拟南芥IDZ蛋白质基序
根据Pfam数据库搜寻结果可得(表2),由结果可知拟南芥IDZ基因家族蛋白含有保守IDZ结构域,该结构域包含一个zf-C2H2_jaz,一个zf_C2H2。zf-C2H2_jaz结构域家族发现于古生菌和真核生物中,jaz含有4个C2H2型锌指基序,它们由长(28~38)氨基酸连接序列连接。jaz在所有被检测的组织中都有表达,并定位于细胞核,主要是核仁。jaz优先结合双链(ds) RNA或RNA/DNA杂交而不是DNA。单个锌指结构域的突变表明,锌指结构域不仅是dsRNA结合的必要条件,也是其核仁定位的必要条件,这表明了依赖于蛋白质的核酸结合能力的复杂贩运机制。此外,jaz可能属于一类以双链RNA结合为特征的锌指蛋白,并可能通过其独特的双链RNA结合特性来调节细胞生长 [17]。
Table 2. IDZ gene family domains in Arabidopsis
表2. 拟南芥IDZ基因家族结构域
3.5. 拟南芥IDZ基因家族的染色体定位
从TAIR数据库上拉取拟南芥IDZ基因家族Genomic Locus相关信息制表(表3),使用MG2C 2.0 (http://mg2c.iask.in/mg2c_v2.0/)导入相关信息绘制可得染色体定位图(图4)。
Table 3. Chromosome distribution information table of IDZ gene family in Arabidopsis
表3. 拟南芥IDZ基因家族染色体分布信息表
IDZ家族染色体定位结果(图4)显示,AtIDZ4、AtIDZ10的基因位于1号染色体,AtIDZ2位于染色体2,AtIDZ1、AtIDZ6、AtIDZ9位于3,AtIDZ8位于4号染色体,AtIDZ3、AtIDZ5、AtIDZ7、AtIDZ11位于5号染色体。基因复制分析结果表明IDZ基因家族没有串联重复基因。

Figure 4. Chromosome mapping of IDZ gene family in Arabidopsis thaliana
图4. 拟南芥IDZ基因家族染色体定位图
3.6. AtIDZ2的蛋白互相作用网络
通过STRING数据库分析AtIDZ2蛋白-蛋白互相作用网络,结果显示节点数为11,如图(图5)所示。其中以AtIDZ2为中心存在多个与之相互作用的蛋白分子,且互作基因的GO基因富集分析结果显示其蛋白涉及的生物学过程多与细胞过程、细胞代谢的调节相关,分子功能多与DNA结合的转录因子活性相关。且这些蛋白多是高迁移率族蛋白(high mobility group protein, HMG蛋白),HMG蛋白在染色质结构与功能及基因表达调控过程中均发挥重要作用,提示AtIDZ家族成员广泛参与生物体生长发育过程中的基因表达调控过程。

Figure 5. AtIDZ2 (IDD5) interacting protein network
图5. AtIDZ2 (IDD5)互作蛋白网络图
3.7. AtIDZ2三维结构分析
AlphaFold产生一个0到100之间的每残留置信评分(pLDDT)。一些pLDDT含量低的区域可能是孤立无结构的。AtIDZ2蛋白三维结构模型见图6。
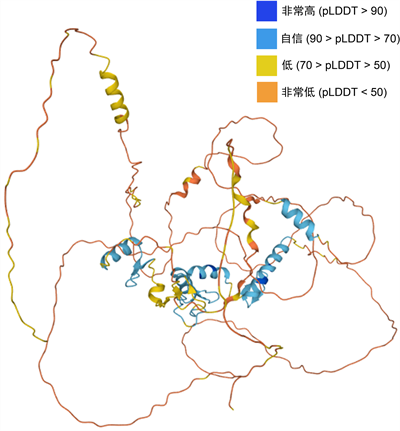
Figure 6. AtIDZ2 protein three-dimensional structure model
图6. AtIDZ2蛋白三维结构模型
4. 讨论与结论
模式生物拟南芥被广泛用于植物生物学各领域研究,因而它的相关生物信息学分析对于植物生物学领域来说是一项非常重要的工作。C2H2型锌指结构蛋白质在植物细胞发育、分化和抑制恶性细胞转化等过程中起着重要作用。本研究通过分析拟南芥IDZ基因家族发现11个家族成员均有保守的IDZ结构域,该结构域包含一个zf-c2h2_jaz,一个zf_C2H2。根据蛋白基序分析可知家族成员蛋白AtIDZ9、AtIDZ10头部和AtIDZ2尾部有一种不保守基序,AtIDZ4、AtIDZ5尾部有另一种不保守基序。由进化树分析可知IDZ家族成员里AtIDZ4的基因与原始基因的差异最小,而AtIDZ2、AtIDZ11的基因与原始基因差异最大。基因家族复制分析结果表明IDZ基因家族没有串联重复基因,且它们多存在于不同染色体上,有发生染色体同片段复制事件的可能性。后续研究选择了IDZ基因家族里的AtIDZ2进行分析,分析结果表明该蛋白与细胞过程、细胞代谢和调节相关,这印证了IDZ结构域含有C2H2型锌指结构域的结果可靠性。本研究为拟南芥IDZ基因家族深入的功能研究奠定了基础。此外,AtIDZ2、AtIDZ4、AtIDZ5、AtIDZ9、AtIDZ10的基序特殊性提示它们可能在进化上具有趋异的功能,有潜在的继续研究价值,相关工作将有利于揭示生物生长发育调控过程的重要机制。