1. 引言
化石燃料是世界能源的主要来源,但是由于化石燃料的不可再生性,导致目前的化石能源面临枯竭,并且引发环境恶化。鉴于这些挑战,人们迫切需要寻找清洁能源来替代传统化石能源。发展绿色能源已经成为我国可持续发展战略的重要组成部分,并受到广泛重视。近年来,随着研究的不断深入,许多能量储存装置得到了长足发展,比如质子交换膜燃料电池、锂离子电池、锌空气电池等。其中质子交换膜燃料电池因其能量密度高、环境友好、能量转换效率高等优点引起研究者的广泛关注 [1] 。质子交换膜燃料电池作为一种优秀的能量储存设备,可以广泛用于交通运输或者便携式电源等领域。
直接甲酸燃料电池(DFAFC)是以甲酸作为燃料,将化学能转化为电能的能量储存装置。虽然甲酸的能量密度低于甲醇,但是DFAFC由于可以使用更薄的质子交换膜和高浓度的甲酸,让DFAFC可以作为较小的电池系统。而且甲酸具有比甲醇更低的理论氧化电势,这会导致电池拥有更高开路电压。直接甲酸燃料电池的工作必须要催化剂的辅助,所以开发合适的阳极甲酸氧化反应(FAOR)的电催化剂是实施DFAFCs技术的关键 [2] 。
直接甲酸燃料电池在阳极处发生甲酸氧化(FAOR),每分子释放出两个电子。阴极发生氧还原反应(ORR),每分子释放出四个电子。直接甲酸燃料电池的反应通常是由贵金属铂基催化剂催化促进的。直接甲酸燃料电池在酸性条件下的反应如下 [2] :
阳极反应:
(1)
阴极反应:
(2)
总反应:
(3)
直接甲酸燃料电池由于其燃料氧化不可避免会产生有毒中间体(一氧化碳),一氧化碳(CO)对质子交换膜燃料电池的阳极催化剂有严重的毒化作用。由于质子交换膜燃料电池的主要成本集中在催化剂(铂基贵金属催化剂)上,所以催化剂被CO毒化对于燃料电池装置来说是致命的。本文的工作是收集并总结近些年直接甲酸燃料电池中Pt基催化剂对CO毒化的缓解策略,并对Pt催化剂后续的抗毒设计提出设想和展望。
2. CO中毒机制
2.1. CO产生机理
可以肯定的是,甲酸在Pt上的电氧化是通过涉及反应中间体和中毒中间体的“双途径机制” [3] 。中毒中间体主要是CO (COad)物种,CO通过自发解离吸附。而且研究发现COad很难除去,除非在远远超过DFAFC中的工作电位(例如,+0.6 V vs. RHE) [4] 。因此,Pt基催化剂在甲酸电氧化过程中容易被COad中毒。直接氧化(途径1)经由脱氢反应发生,而不形成COad:
途径1:
(4)
第二反应途径(途径2)通过脱水形成吸附的一氧化碳(CO)作为反应中间体:
途径2:
(5)
在反应途径2中,甲酸首先吸附到催化剂表面上,形成中间吸附的CO物种,然后将其氧化成气态CO2终产物 [5] 。对于直接甲酸燃料电池,脱氢是所需的反应途径,以提高整体电池效率并避免催化剂中毒。选择阳极催化剂以通过反应途径1引导甲酸氧化是关键的。
2.2. CO对Pt的毒化机理
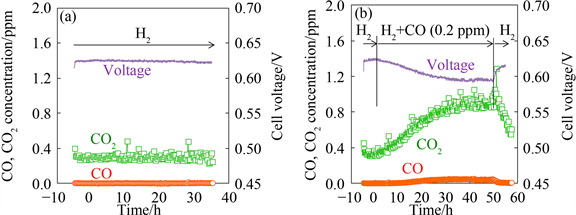
Figure 1. Variations of cell voltage, CO2, and CO concentrations under (a) high-purity hydrogen and (b) CO (0.2 ppm) contaminated hydrogen [6]
图1. 电池电压、CO2和CO浓度在(a) 高纯度氢气和(b) CO (0.2 ppm)污染的氢气下的变化 [6]
Matsuda [6] 等人为深入研究CO对PEMFC的具体影响,用商业Pt/C安装了一块PEMFC装置,实验结果表明在0.2 ppm的CO中,电池的稳态电位降低了29 mV。如图1所示,在通入CO后,CO2的浓度显著上升,电池电压明显下降。50小时后再次通入高浓度的氢气后电池电压再次恢复。Chen [7] 等人的研究表明随着CO浓度的提高,PEMFC的电压损失会大幅增加,从而极大限制了电池的输出功率。因此,CO对于燃料电池的负面影响非常令人担忧。
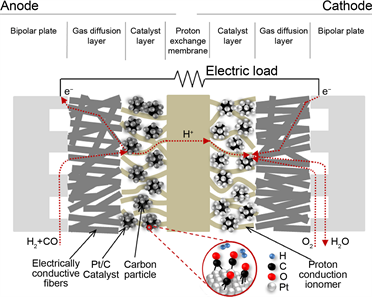
Figure 2. The role of CO on the anode catalyst [7]
图2. CO在阳极催化剂上的作用 [7]
在一般阳极反应下,CO与H竞争铂上的活性位点(如图2)。这是因为CO比H2更容易吸附在Pt表面(CO在Pt上的吸附热为134 kJ·mol−1,H2为87.9 kJ·mol−1)。这种行为导致甲酸氧化反应(FAOR)可用的活性点位减少。而CO在铂的表面聚集,因此CO比甲酸更容易吸附在Pt的表面,导致FAOR可用的表面减少,从而限制了其性能。从传质的角度看,当CO吸附在阳极催化剂上时,会抑制氢解离成质子和电子,因此CO对使用Pt和Pd作为电催化剂的DFAFC的功率输出的影响随着浓度的增加而增加,这种影响在低温下尤为显著 [8] [9] [10] 。
(6)
(7)
(8)
CO在铂表面上的覆盖达到阈值,CO的电氧化便会发生。对于纯铂电极,其发生电位高于0.6 V (vs. RHE) [11] 。图3呈现了在23℃下在Pt/C催化剂上CO氧化的循环伏安图 [1] 。当电位达到超过一定阈值,就会允许CO进行氧化,出现一个突出的电流,在图3的循环伏安图中呈现出一个明显的峰,即CO氧化峰。

Figure 3. Cyclic voltammograms of CO on polycrystalline Pt at 23˚C [1]
图3. 在23℃下CO在多晶Pt上获得的循环伏安图 [1]
3. 抗CO催化剂的研究现状
目前,能源市场主要由化石能源和以锂电池为主的电池技术占据主导地位。尽管直接甲酸燃料电池在性能上具有巨大的应用潜力,但它仍然面临一些关键问题,这些问题严重限制了其发展:
1) 催化剂的成本高昂:在直接甲酸燃料电池中,催化剂的成本占是整个电池装置的很大比例。 [12] 大量的研究表明,Pt族金属是催化甲酸氧化性能最出色的材料,然而,Pt属于贵金属,所以在催化剂中降低Pt的载量是降低催化剂成本的关键。
2) 催化剂易被毒化:在直接甲酸燃料电池运行过程中,甲酸燃料分子断裂不可避免的会产生CO中间体,CO对Pt的毒化机理在本文的上一部分已经阐述。因此,提高催化剂的抗CO中毒性能成为催化剂设计的重要方向。这可以通过改进催化剂的结构或引入新的催化剂材料来实现,以减轻CO对催化剂的不利影响。
3.1. 单原子催化剂
单原子催化剂是只含有分散在载体上的孤立单个原子的催化剂,单原子催化剂可以最大限度的将活性位点暴露出来。大量的研究证明,FAOR的反应途径(双途径机制)可以通过改变Pt基催化剂的整体尺寸调节。Pt的大团簇催化剂更容易发生FAOR中的间接途径,而直接途径更容易发生在被孤立的单原子上。 [13] 因此单原子催化剂更容易避免产生CO中间体,从而提高催化剂的抗CO毒化性能。
众所周知,Pt-Pt键的键能较高 [1] ,所以构建Pt单原子催化剂具有一定的挑战。当衬底上的Pt负载量很低时,在Pt生长的早期阶段会形成孤立的Pt单原子。 [14] 例如Au@Pt核壳结构催化剂是在Au衬底上生长Pt,但是Pt在衬底上的覆盖率不到10%,由于Pt的稀疏,同样导致了催化剂的成本上升。Liu [13] 等人发现了一种新的配体介导的生长机制,可以用湿法化学法构建单原子Pt。该方法的关键是在合成体系中亚硫酸盐(
)配体,该配体在Pt位点上有较强的吸附作用,而在Au位点上的吸附较弱。由于在预先存在的Pt位点上沉积Pt原子涉及到从Pt位点上先解吸预吸附的
配体,Pt-Pt键的形成遇到了一个高能垒,与Au-Pt键的形成相比,Pt-Pt键的形成是一个能量上不利的过程(见图4)。所以在Pt位点上Pt的生长是一个自终止的过程,导致大量的单原子Pt形成。Liu [13] 等人利用Ag@Au核壳纳米片作为衬底,在此衬底上可以更好的形成Pt(111)面,而且单原子Pt在Ag@Au核壳纳米片上有较高的负载量。图5(a)、图5(b)展示了Ag@Au@PtnL (n为Pt的覆盖率)纳米片的典型高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像和高分辨率透射电子显微镜(HRTEM)图像。图5(c)是Ag@Au@Pt纳米片的X射线能谱仪(EDS)图,清晰的显示出Pt均匀的分散在Ag@Au纳米片衬底的整个表面。
Ag@Au@PtnL在电化学测试中展现出卓越的甲酸氧化性能(如图5(d),以商业Pt/C为基准),图中可以看出当Pt的覆盖率在0.43~1.3时,在电位0.96 V左右有明显的CO氧化峰(图5(d)中黑色箭头指向),而当Pt的覆盖率0.26或更小时,几乎无法观察到CO氧化峰。这表明反应路径从间接向直接路径转换。图5(e)中表明随着Au衬底上Pt原子层的减少,j0.96 V/j0.57 V值连续降低,进一步证实了由于反应路径的转换而导致FAOR中CO中毒程度降低。这意味着当Pt的覆盖率较高时,容易形成Pt的团簇。单原子Pt催化剂在电催化的过程中展现出很高的耐久性,经过3600秒的计时电流测试后(i-t)仍然保持着很高的电流密度,并且是商业Pt/C的1270倍。如此高的耐久性可以归因于单原子Pt孤立的沉积在Au衬底上,极大促进了FAOR通过直接途径进行,使催化剂的CO中毒被很有效的抑制。大量的单原子催化剂的研究表明,当单原子Pt颗粒尺寸减小到原子水平是提高金属催化剂利用率的有效途径,这不仅最大化了金属的原子效率。单原子催化剂目前面临的问题是当金属粒子的尺寸减小到原子级时,金属粒子的表面自由能急剧增加导致原子形成团簇。目前,更多的研究人员倾向于将Pt单原子与电负性强的元素(如N、S、O)进行配位,可以有效提高单原子的稳定性。
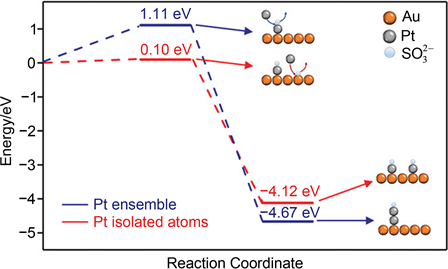
Figure 4.
ligands participate in the growth of Pt atoms adsorbed on pre-existing Au sites to form Au-Pt bonds and thus form Pt single atoms (red lines) as well as pre-existing Pt sites on Au substrates to form Pt-Pt Schematic diagram of the energy evolution when bonding and thus forming Pt clusters (blue lines) [13]
图4. 当
配体参与Pt原子吸附在预先存在的Au位点的生长以形成Au-Pt键并因此形成Pt单原子(红线)以及在Au衬底上的预先存在的Pt位点形成Pt-Pt键并因此形成Pt团簇(蓝线)时的能量演化示意图 [13]
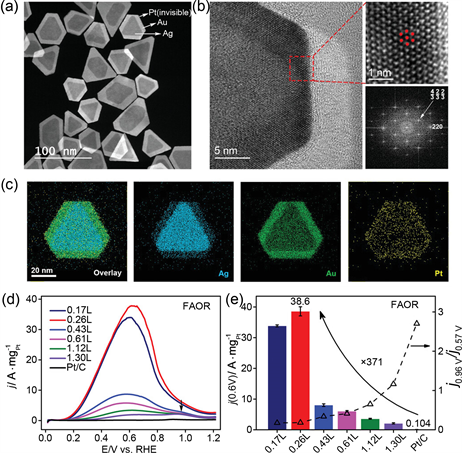
Figure 5. Characterization and electrochemical testing of Ag@Au@PtnL: (a) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM); (b) High-resolution transmission electron microscopy (HRTEM) image (Pt: 26%); (c) Energy-dispersive X-ray spectroscopy (EDS) image of Ag, Au, and Pt in Ag@Au@PtnL nanoplates; (d) Cyclic voltammetry (CV) diagrams of catalysts with different Pt contents in N2-saturated 0.5 M H2SO4 + 0.25 M HCOOH at a scan rate of 50 mV·s−1, the arrow points to the indirect path of FAOR CO oxidation peak; (e) Left axis: comparison of mass activities of catalysts in FAOR; right axis: j0.96 V/j0.57 v of catalysts [13]
图5. Ag@Au@PtnL的表征和电化学测试:(a) 高角度环形暗场扫描透射电子显微镜(HAADF-STEM);(b) 高分辨率透射电子显微镜(HRTEM)图像(Pt: 26%);(c) Ag@Au@PtnL纳米板中Ag、Au和Pt的X射线能谱仪(EDS)图;(d) 不同Pt含量的催化剂在N2饱和的0.5 M H2SO4 + 0.25 M HCOOH中在50 mV·s−1的扫描速率下的循环伏安(CV)图,箭头指向是由于FAOR的间接路径导致的CO氧化峰;(e) 左轴:FAOR中催化剂的质量活性的比较;右轴:催化剂的j0.96 V/j0.57 v [13]
3.2. 合金催化剂
近年来,对纳米合金催化剂的研究越来越多,已经表现出可靠的催化活性和良好的抗CO中毒性能。然而,纳米颗粒催化剂在循环中的差的粘附性和极容易发生团聚通常导致其催化性能的严重退化,从而严重限制了DFAFC的商业化。此外,纳米颗粒合成程序的尺寸敏感性和复杂性也限制了纳米颗粒催化剂制造的规模扩大,这也阻碍了它们的工业应用。必须找到不仅具有高活性(如纳米颗粒),而且还具有耐久性的催化剂,使其在长期测试和应用中可行。目前对于Pt基催化剂的改性方法有很多,其中最普遍的是与其他金属进行合金化。合金化不仅可以减少Pt的含量,降低成本,而且可以通过调节Pt的电子结构起到提高Pt的抗CO性能的作用,比如添加Bi和Co可以提高催化剂对OHads的吸附,从而加快CO氧化。 [15] 合金催化剂大致可以分为二元合金催化剂、三元合金催化剂和四元甚至高熵合金催化剂。
3.2.1. 二元合金催化剂

Figure 6. Morphology, structure and composition characterization of ultrathin AgPt nanowires prepared by Jiang [16] et al.: (a) and (b) are typical TEM images at different magnifications; (c) is a high-resolution TEM image of the material, and the inset is the corresponding selected electron diffraction image; (d) Elemental map of the material
图6. Jiang [16] 等人所制备的超薄AgPt纳米线的形貌、结构和组成表征:(a)和(b) 是以不同放大倍数的典型透射电镜图像;(c) 是该材料的高分辨率透射电镜图像,插图为相应的选取电子衍射图像;(d) 该材料的元素分布图
目前为止,研究者们已经进行了大量关于直接甲酸燃料电池的二元Pt基催化剂的研究,例如PtAg [16] 、PtAu [17] 、PtNi [18] 、PtMn [19] 、PtCo [20] 、PtPb [21] 、PtSn [22] 等。大量的研究表明,一维结构的纳米线具有独特的特征,具有稳定的结构,在电催化过程中不易被溶解,还可以暴露出更多的表面原子并提供丰富的活性位点,这有利于增强材料的电催化活性。 [23] [24] 受益于其各向异性,电子的传输也得到了增强。Jiang [16] 等人通过一种简单的策略合成了一种一维超薄的PtAg纳米线,此方法同时结合了合金和一维材料的优势。图6(a)、图6(b)分别展示了PtAg纳米线的低倍和高倍下的透射电镜(TEM)图像,可以明显看出,该产物是由许多具有波状结构和高度均匀的一维纳米线组成,而且说明纳米线的产率很高(没有其他形态的产物出现)。由于其出色的柔性,纳米线彼此之间容易相互交织形成纳米线网格。观察图6(b)可以表明每个纳米线的长度在数十纳米的范围内,经过测量平均直径为3.9 nm左右。图6(c)显示了单个纳米线具有粗糙的表面和不均匀的直径,表明它基本上是由沿着不同方向互连的单个纳米晶体构成的。因此,纳米线呈现波浪形状,并且在相邻纳米晶体发生交织的地方可以容易地观察到明显的缺陷(如图6(c)中红色箭头所示),材料中的缺陷可以才催化期间充当高活性位点,有效的提高材料的催化性能。高分辨率透射电镜图(如图6(c))中观察到清晰的晶面间距(0.232 nm),原因是存在AgPt (111)面。该晶面间距值介于面心立方相Pt (0.227 nm)和Ag (0.236 nm)的(111)晶格间距之间,意味着所制备的超薄AgPt纳米线的合金化组成。图6(c)的插图证明了纳米线具有多晶结构。在元素图谱分析(图6(d))图中清楚的表明Ag和Pt重叠分布,进一步证明了产物由AgPt合金组成。
为了验证AgPt超薄纳米线的电催化性能,对材料进行甲酸氧化的电化学性能测试。在30℃、0.5 M H2SO4 + 1.0 M HCOOH溶液中以50 mV·s−1的扫描速率进行甲酸的电氧化,其性能与商业Pt黑和AgPt纳米颗粒进行了比较。图7(a)展示了在N2饱和的0.5 M H2SO4溶液中记录的三种催化剂的典型循环伏安图(CV)曲线。所有催化剂在0.02 V~0.32 V区域内显示出明显的与质子还原/氢氧化相关峰。该电位区域可以计算出超薄AgPt纳米线的电化学活性面积(ECSA)为15.8 m2·g−1,略小于AgPt纳米颗粒(17.6 m2·g−1)和商业Pt黑催化剂(20.8 m2·g−1)。出现这种情况的原因是Ag原子的掺杂一定程度上降低了表明Pt的含量,此外由于其一维的形貌结构也是导致电化学活性面积(ECSA)低的原因[25]。图7(b)显示了三种催化剂FAOR的循环伏安(CV)图。三种催化剂的正向扫描的区域均有两个明显的阳极峰,峰I位于0.57 V左右处,对应于甲酸的直接氧化途径(
);峰II位于0.92 V~0.99 V范围内,对应于甲酸氧化的间接途径(
),表明在此电位上产生了CO有毒中间体。通过比较三种催化剂的CV图,发现超薄AgPt合金纳米线FAOR起始电势比AgPt纳米颗粒和商业Pt黑催化剂的起始电势显著更负,表明FAOR更容易在AgPt合金纳米线上发生。仔细观察CV图可以发现AgPt合金纳米线的峰I和峰II面积比(R)为2.55,远大于AgPt纳米颗粒和商业Pt黑,如此高的R值归功于AgPt合金纳米线在FAOR中主要实现直接氧化途径。如图7(c)中的R值和周转频率(TOF)图,这两个数值都能体现AgPt合金纳米线具有优秀的电催化效率和电流密度,进一步证明了超薄AgPt合金纳米线展现了令人兴奋的电催化活性。
图7(e)展示了三种催化剂的塔菲尔(tafel)图,通过外推Tafel曲线,计算出超薄AgPt合金纳米线的交换电流密度(j0)为0.17 mA·cm−2,分别比AgPt纳米颗粒(0.057 mA·cm−2)和商业Pt黑催化剂(0.025 mA·cm−2)大3.1倍和6.8倍。因此,超薄AgPt合金纳米线的电荷转移电阻(Rct)比商业Pt黑催化剂的低得多,同时Rct值的降低表明超薄AgPt合金纳米线表面可以有效去除CO有毒中间体 [26] 。研究人员对超薄AgPt合金纳米线进行了稳定性测试,通过在含有1.0 M HCOOH的0.5 M H2SO4溶液中的0.57 V下进行计时电流法(CA)测量3000秒。如图7(f)所示,超薄AgPt合金纳米线相比于其他两种催化剂的电流衰减更缓慢,表明超薄AgPt合金纳米线具有更高的稳定性。
Jiang [16] 等人的合成的超薄AgPt合金纳米线是甲酸燃料电池中典型的二元合金催化剂,研究人员通过控制特定的形貌有效提升缺陷数量,从而使催化剂对FAOR活性增强,同时有效控制反应途径,减少CO的产生。从根源上减少CO有毒中间体,因此催化剂可以展现出优异的CO耐受性。Stevanovic [22] 等研究者通过多元醇合成法成功制备了稳定的PtSn纳米颗粒,并负载在碳上,制备了PtSn/C催化剂。如图8(a)示,说明向Pt中添加Sn可以显著增强FAOR的活性。当Sn存在于表面上时,对FAOR的改进主要是防止COad阻挡表面Pt原子。 [24] 在图8(b)中氧化去除COad已经被证实发生在较低的电位,然后显着的活性增强在高电位区域。DFT结果表明,PtSn/C在低电位区的COad氧化是双功能机制的结果。根据这种机制,OH物种形成在强吸附的Sn原子,SnO2氧化物或合金Sn上。经过研究发现在CO是Pt中毒过程中,Sn可以在低电势下提供OH。所以说催化剂表面的Sn对CO在低电位下氧化起到了促进作用。

Figure 7. [16] Formic acid (HCOOH) oxidation reaction (FAOR) activity of as-prepared ultrathin AgPt alloy nanowires, AgPt alloy nanoparticles, and commercial Pt black catalysts: (a) CV curves in N2-saturated 0.5 M H2SO4 solution; (b) ECSA-normalized FAOR CV curves were recorded in N2-saturated 0.5 M H2SO4 + 1.0 M HCOOH solution; (c) The ratio of peak I to peak II and the transition frequency (TOF); (d) Specific activity and mass activity; (e) Tafel diagram; (f) Chronoamperometry (CA) curves recorded at 0.57 V
图7. [16] 制备的超薄AgPt合金纳米线、AgPt合金纳米颗粒和商业Pt黑催化剂的甲酸(HCOOH)氧化反应(FAOR)活性:(a) 在N2饱和的0.5 M H2SO4溶液中CV曲线;(b) 在N2饱和的0.5 M H2SO4 + 1.0 M HCOOH溶液中记录ECSA归一化的FAOR CV曲线;(c) 峰I与峰II的比率和转换频率(TOF);(d) 比活度和质量活度;(e) Tafel图;(f) 在0.57 V下记录的计时电流法(CA)曲线
Waenkaew [27] 等研究者通过2-丙烯酰胺基-2-甲基-1-丙烷磺酸(PAMPs)改性的氧化石墨烯(GO)作为载体材料(PAMPs/Go),并采用电沉积法制备了双金属PtPd催化剂。图8(c)显示了催化剂在0.5 M H2SO4 + 0.5 M HCOOH溶液中的CV。在阳极扫描过程中观察到两个甲酸氧化的特征峰,分别位于0.34 V和0.68 V。第一个峰与甲酸通过脱氢氧化的直接氧化相关,涉及消除两个氢原子以形成CO2。第二个峰涉及经由脱水反应的间接氧化,因为来自甲酸的“非法拉第”解离的预吸附CO (COads)被氧化。对于反向扫描,0.35 V处氧化峰对应于从正向扫描中发生的中间体的去除。3Pt3Pd/10% PAMPs/GO相比Waenkaew等人制备的其他催化剂有更好的比活性和质量活性。从图中可以很明显的分析出,PAMP不仅增加了电流密度,而且增加了耐CO的催化剂的耐久性。图8(d)是几种材料在0.5 M H2SO4 + 0.5 M HCOOH溶液中塔菲尔(Tafel)图,其中3Pt3Pd/10% PAMPs/GO的Tafel斜率相对较低,表明其有具有快速的电子传输性能和电荷转移动力学,验证了催化剂具有很好的电催化甲酸氧化活性。如图8(e)是对3Pt3Pd/10% PAMPs/GO进行的CO耐受性测试,CO氧化峰在第一次扫描中出现,第二次扫描中CO氧化峰消失,说明CO在第一次扫描后被完全氧化。图中的CO氧化起始电位相比其他催化剂低,说明其具有更好的CO耐受性。这种材料有效的抗毒性能得益于Pt和Pd原子的结合而改变的Pt的电子结构减弱了CO和Pt的相互作用,导致弱键合的COads部分的氧化 [28] 。
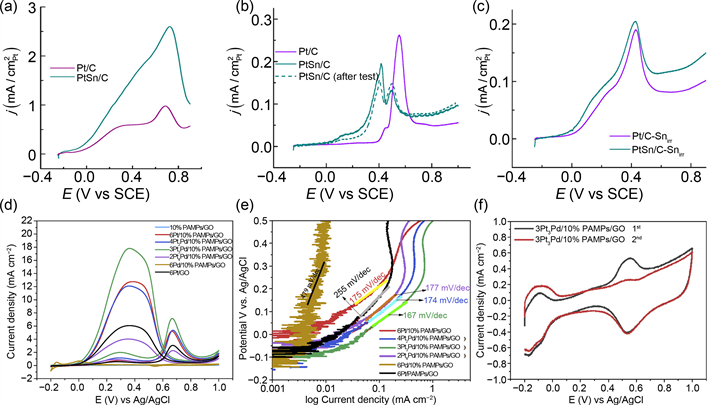
Figure 8. Commercial Pt/C and PtSn/C under the condition of 0.1 M HClO4, v = 50 mV·s−1, (a) The potential kinetic curves of 0.5 M HCOOH in the extension potential region; (b) COad stripping curve; (c) COad stripping voltammetry curves of Pt/C-Snirr and PtSn/C-Snirr in 0.1 M HClO4, v = 50 mV·s−1 after Sn modification on the catalyst surface after annealing treatment [25] ; (d) The prepared catalyst was in 0.5 M H2SO4 + 0.5 M HCOOH solution with a scan rate of 50 mV·s−1 and a potential range of 0.2 V to 1.0 V; (e) Tafel diagram of the catalyst in 0.5 M H2SO4 + 0.5 M HCOOH; (f) CO stripping voltammograms of 3Pt3Pd/10% PAMPs/GO were obtained in 0.5 M H2SO4 [28]
图8. 商业Pt/C和PtSn/C在0.1 M HClO4, v = 50 mV·s−1的条件下,(a) 0.5 M HCOOH的延伸电位区电位动力学曲线;(b) COad剥出曲线;(c) 经过退火处理在催化剂表面修饰Sn后的Pt/C-Snirr和PtSn/C-Snirr在0.1 M HClO4,v = 50 mV·s−1中的COad溶出伏安曲线 [25] ;(d) 制备的催化剂在0.5 M H2SO4 + 0.5 M HCOOH溶液中,扫描速率为50 mV·s−1,电位范围为0.2 V~1.0 V;(e) 0.5 M H2SO4 + 0.5 M HCOOH中催化剂的Tafel图;(f) 在0.5 M H2SO4中获得了3Pt3Pd/10% PAMPs/GO的CO溶出伏安 [28]
3.2.2. 多元合金(三元及以上)催化剂
Xie [29] 等人使用电沉积的方法制备一种超低Pt负载纳米多孔Au-Cu-Pt合金薄膜,然后通过去合金化去除Cu,最终合成出用于FAOR的超低Pt负载的三金属纳米多孔(np) Au-Cu-Pt催化剂。为了进一步深入研究np Au-Cu-Pt (2.6%)膜中的Pt活性面积,采用CO剥离法测量。如图9(a)所示,GC上Pt的CO氧化电流(黑线)显著高于np Au-Cu-Pt (2.6%)催化剂的CO氧化电流(红线),这证实了在纳米多孔合金表面上存在非常小的Pt部分。图9(b)展示了在np Au-Cu-Pt (2.6%)上的CO剥离实验的第一和第二阳极扫描。结果清楚地表明,CO在第一次扫描中被完全氧化,使得后续扫描(第二次循环)的电流仅与np Au-Cu-Pt的背景信号(2.6%)相关。
这种Au-Cu-Pt合金薄膜也具有很强的甲酸氧化性能。研究人员在0.1 M HClO4 + 2 M HCOOH溶液中通过循环伏安法(CV)测试其性能,如图9(c)所示。这是沉积在玻碳电极上的普通Pt膜的465倍。循环测试表明,较低的Pt负载量(1.3%)可以将质量活性提高至
,如图9(d)所示。在这种情况下,不仅质量活性与现有技术的Pt单原子催化剂 [30] 相当,而且Pt负载量较低的催化剂np Au-Cu-Pt (1.3%)同时保留了无CO中毒类型的CV曲线。
研究者对Au-Cu-Pt合金薄膜的稳定性进行了长期循环测试,结果表明较厚的合金薄膜(80 nm)的催化活性可以保持超过2100次循环,而较薄的薄膜(33 nm)在经过1200次循环后就失去很大一部分活性,出现此现象的原因可能是厚度不同的催化剂Pt的总量不同。在np Au-Cu-Pt (2.6%)催化剂的2200次循环测试后进行的催化剂元素含量的EDS评价表明Cu的大量损失。这表明,虽然最初存在于合金中,但Cu也在支持无钝化行为中起重要作用。此外,作为活性金属的Pt的损失也有导致FAOR中的总体活性损失。由于配体效应,合金催化剂的表面电子结构与纯铂催化剂相比发生了变化。纽曼 [31] 等人的研究表明,np Au-Ag-Pt合金的韧带表面主要由Au和Pt构成。Au和Pt之间的合金化已显示导致Pt的d带中心从−2.25 eV大幅上移至−1.82 eV,这非常接近Pd的d带中心−1.83 eV。众所周知,由于其d带中心位置,Pd提高了FAOR通过直接途径反应的几率 [32] [33] 。在此工作中,由于Au-Pt合金化导致的d带中心偏移可以通过图9(a)中所见的CO氧化峰位置来证实。图9(e)中所示的Au、Cu和Pt之间的协同效应可以促进FOAR过程,如果与Au形成合金化的Pt可以破坏C-H键,而周围的Au原子可以将-COOH转化为CO2 [34] 。此外,早先已经发现Cu原子用作含氧物质的位点,从而促进去除中毒中间体如OHad和COad。 [35] 表面Cu原子可以承担大部分OHad氧化,从而防止Pt氧化的发生。随着Cu在循环期间的损失,催化剂耐受性降低。
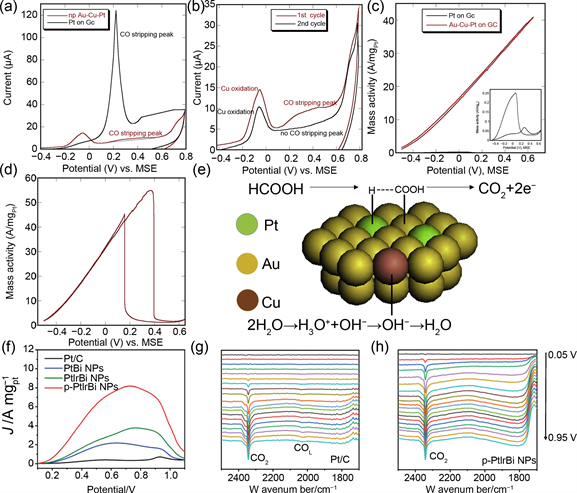
Figure 9. (a) CV curves of Pt on GC (black) and np Au-Cu-Pt (red, 2.6%) in 0.1 M HClO4 solution, and (b) first ( Red) and CV curves of the second (black) cycle, scan speed: 50 mV·s−1. CV curves show that the activities of np Au-Cu-Pt are (c) 2.6% and (d) 1.3% when loaded with Pt in 0.1 M HClO4 + 2 M HCOOH solution, respectively. Activity of Pt films on GC; (e) Synergistic effect on the FAOR of np Au-Cu-Pt with highly dispersed surface Pt [31] ; (f) Anodic scanning polarization curves of different catalysts in 0.1 M HClO4 + 0.5 M HCOOH solution. In situ infrared spectroscopy (FTIR) at 0.05 V to 0.95 V was recorded on (g) commercial Pt/C and (h) p-PtIrBi NPs/C catalysts [36]
图9. 0.1 M HClO4溶液中(a) Pt在GC (黑色)和np Au-Cu-Pt (红色,2.6%)上的CV曲线,和(b) Au-Cu-Pt (2.6%)上的第一(红色)和第二(黑色)循环的CV曲线,扫描速度:50 mV·s−1。CV曲线显示,在0.1 M HClO4 + 2 M HCOOH溶液中,负载Pt时,np Au-Cu-Pt的活性分别为(c) 2.6%和(d) 1.3%。Pt薄膜在GC上的活性;(e) 协同效应对表面Pt高度分散的np Au-Cu-Pt的FAOR有影响 [31] ;(f) 不同催化剂在0.1M HClO4 + 0.5 M HCOOH溶液中阳极扫描极化曲线;(g) 商用Pt/C;(h) p-PtIrBi NPs/C催化剂上记录了0.05 V~0.95 V的原位红外光谱(FTIR) [36]
Sun [36] 等研究者通过设计一种新型的二维多孔PtIrBi纳米板三金属催化剂(p-PtIrBi NPs),引入铱(Ir)有助于促进甲酸氧化,特别是通过脱氢途径,从而提高催化剂对CO的耐受性。在实验中实现了这一策略。优化后的p-PtIrBi NPs/C具有8.2 A·mg−1的高质量活性和55.9%的稳定性,是迄今为止报道的最好的甲酸氧化催化剂之一,远高于PtIrBi NPs/C、PtBi NPs/C和Pt/C。CO氧化和原位傅里叶变换红外(FTIR)实验共同证明,“Pt-Bi”和“Ir-Bi”这两种类型的适当位点赋予催化剂抑制CO中毒的特性,从而实现甲酸氧化反应的超高活性和稳定。所有制备的催化剂(p-PtIrBi NPs/C、PtIrBi NPs/C和PtBi NPs/C)对FAOR的起始电位都比商用Pt/C催化剂低得多。最活跃的p-PtIrBi NPs/C的起始电位值为116 mV,比Pt/C低240 mV (如图9(f))。图9(f)、图9(g)显示了在0.05 V~0.95 V范围内获得的商业Pt/C、PtBi NPs/C和p-PtIrBi NPs/C的原位傅里叶变换红外光谱(FTIR)。在p-PtIrBi NPs/C的红外光谱中,CO2出现在0.1 V的低电位,低于PtBi NPs/C (0.15 V)和Pt/C (0.3 V),表明该Ir位点有利于HCOOH在p-PtIrBi NPs/C上的直接氧化。p-PtIrBi NPs/C中无CO谱,表明其对CO中毒具有较高的抗性,且反应过程以脱氢途径为主。
类似的,三元催化剂还有Bhalothia [37] 等人合成的PdRuPt纳米催化剂、Qin [38] 等人Pt-Fe-Mn三元高指数晶面催化剂等都展现出不俗的甲酸氧化活性和CO耐受性。高熵合金催化剂是目前比较热门的催化剂形态,但在用于甲酸氧化的Pt基高熵合金催化剂却屈指可数。高熵合金可以通过廉价的金属掺杂降低催化剂的成本,多金属掺杂可以极大可能调节Pt的d带中心,增加晶格缺陷,改善金属分子之间的键合,从而提高催化剂的催化活性和CO耐受性 [39] 。Pt与过渡金属的合金化有利于电子的获得,从而有效地提高了催化剂的性能。合金的加入降低了Pt的d带中心,优化了电催化剂的性能。大量的研究表明,过渡金属或贵金属与Pt的合金化通过压缩应变和电子效应有效提高催化剂的CO耐受性。
3.3. 异质结构催化剂
异质结构催化剂是一种混合材料,通过多种固体材料的组合从而形成各组分之间的界面,从而形成一种协同效应,使催化剂具有优越的性能,并在催化应用中具有不错的潜力。 [40] [41] 大量研究表明,二维核/壳纳米结构催化剂,在提高性能和寿命方面有一定优势,但是很少对核壳结构的甲酸氧化路径有研究。
Hu [42] 等人合成了一种新颖且表面不均匀的PtPbBi/PtBi核/壳纳米片(PtPbBi/PtBi NPs)作为高活性和选择性的FAOR催化剂,此纳米板由一个内部的PtPbBi核和一个厚度不均匀的PtBi壳组成。PtBi和PtPbBi之间存在局域电子相互作用,同时高耐受性的PtBi壳层可以有效抑制CO的生成/吸附,导致FAOR完全存在脱氢途径。PtPbBi/PtBi核/壳纳米片表现出前所未有的甲酸氧化催化活性,质量活性和比活性也远高于商业Pt/C。因为不均匀的壳层可以有效地为FAOR过程提供更多的PtBi活性位点(如图10(b))。由于表面结构不规则,PtPbBi/PtBi NPs/C比PtPb/Pt NPs/C具有更大的ECSA。从不同催化剂在0.5 M H2SO4 + 0.5 M HCOOH溶液中的正向扫描曲线(图10(c))可以看出,PtPbBi/PtBi NPs/C具有很高的催化活性,而且PtPbBi/PtBi NPs/C仅在低电位处出现强峰,这与HCOOH直接氧化生成CO2的反应规律一致,说明甲酸氧化反应主要通过脱氢途径进行。研究人员通过X射线光电子能谱仪(XPS)对材料的元素价态进行分析,研究发现PtPbBi/PtBi NPs/C中Pt的结合能(BE)发生负偏移,这一现象证明电子偏移能够增强质子结合和脱氢过程,同时削弱催化剂表面对有害吸附中间体的吸附强度,从而提高了催化剂对CO的耐受性。
Al-Qodami [43] 等研究人员设计了一种铁和镍纳米线氧化物,使得其串联在裸铂基底上形成的三元催化剂(FeOx/NiOx/Pt)。FeOx/NiOx/Pt催化剂的催化活性和CO耐受性分别比纯Pt催化剂提高了4.8倍和1.6倍。研究人员在实验过程中发现,在0.2 M NaOH溶液中,FeOx/NiOx/Pt催化剂在0.5 V电位活化下,Fe2+/Fe3+转化成功地缓解了Pt基催化剂的永久性CO中毒。有意思的是,经过活化的FeOx/NiOx/Pt催化剂活性比纯Pt催化剂高7倍。在低电位下(<0.25 V)下,直接途径机制占主导地位。图10(e)是研究者制备的催化剂在碱性条件下的催化活性对比,FeOx/NiOx/Pt的催化活性优于其他催化剂。在CO溶出实验中,FeOx/NiOx/Pt和a-FeOx/NiOx/Pt电流的峰值强度分别为1.4和1.2 mA·cm−2,远低于其他催化剂。这表明FeOx/NiOx/Pt和a-FeOx/NiOx/Pt催化剂具有较高的CO耐受性。异质结构催化剂得益于其特殊的形貌结构,在其边界或表更容易暴露活性位点,使其具有不错的催化效率。此外异质结构催化剂对于甲酸的催化主要集中在直接途径上,从根源上减少CO的析出,对催化剂的抗毒性能起到关键作用。因此异质结构的催化剂是一种很有前景的材料。
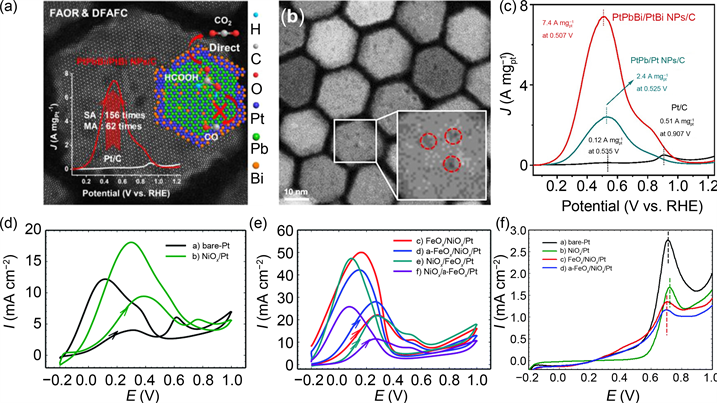
Figure 10. (a) SEM image and structure diagram of PtPbBi/PtBi core/shell nanoplates; (b) Enlarged HAADF-STEM (High Angle Annular Darkfield Scanning Transmission Electron Microscopy) image. The inset shows a magnified image of a single PtPbBi/PtBi NP; (c) Forward scan curves of different catalysts for FAOR in 0.5 M H2SO4 + 0.5 M HCOOH electrolyte at a scan rate of 50 mV·s−1 [42] ; (d) CVs of pure Pt, NiOx/Pt, FeOx/NiOx/Pt, a-FeOx/NiOx/Pt; (e) FAOR of pure Pt, NiOx/Pt, FeOx/NiOx/Pt, a-FeOx/NiOx/Pt; (f) CO oxidation of pure Pt, NiOx/Pt, FeOx/NiOx/Pt, a-FeOx/NiOx/Pt electrodes in 0.5 M H2SO4 at 50 mV·s−1 [43]
图10. (a) PtPbBi/PtBi核/壳纳米板的SEM图像以及结构图;(b) 放大的HAADF-STEM (高角度环形暗场扫描透射电子显微镜)图像。插图显示单个PtPbBi/PtBi NP的放大图像;(c) 在0.5 M H2SO4 + 0.5 M HCOOH电解液中,扫描速率为50 mV·s−1时,不同催化剂对FAOR的正向扫描曲线 [42] ;(d) 纯Pt、NiOx/Pt、FeOx/NiOx/Pt、a-FeOx/NiOx/Pt的CV;(e) 纯Pt、NiOx/Pt、FeOx/NiOx/Pt、a-FeOx/NiOx/Pt的FAOR;(f) 在0.5 M H2SO4中,在50 mV·s−1下,纯Pt、NiOx/Pt、FeOx/NiOx/Pt、a-FeOx/NiOx/Pt电极的CO氧化 [43]
4. 总结与展望
直接甲酸燃料电池(DFAFCs)拥有安全便携、环境友好等优点,被研究人员寄予厚望。但是直接甲酸燃料电池受限于Pt催化剂的价格高昂、易被毒化失活等问题,限制了其商业化发展。经过研究人员近年来对阳极催化剂的不断改进,有效地降低了催化剂的成本、提高催化活性以及CO耐受性等。尽管如此,直接甲酸燃料电池距离商用依然有一定的距离,这是因为目前的催化剂总体成本偏高、催化剂无法实现大规模化,制备技术尚需开发。未来催化剂的设计工作可以按照以下几个方面来进行:高熵合金因其具有多元性和高度可调性,可以被设计为DFAFCs耐CO催化剂,并且有大量可调节和改性空间没有被发掘。目前催化剂的载体较为单一,限制了催化剂的发展,石墨烯、碳纳米管等新型碳材料具有特殊的结构,这类材料对催化剂的影响有待进一步研究。
NOTES
*通讯作者。