1. 引言
酰胺化合物广泛存在于天然产物中,是蛋白主的重要组成部分 [1] [2] 。酰胺在许多合成药物、医药和精细化学品的合成中有着广泛的应用 [3] [4] ,据统计约有25%的药物分子中含有酰胺官能团,因此对酰胺化合物的合成研究具有重要的意义 [5] 。实验室酰胺化合物的合成方法有很多。传统酰胺化合物的合成是由羧酸或者羧酸衍生物与胺反应合成 [6] [7] [8] 。近年来随着新型催化剂的应用和新方法的研究,酰胺化合物的合成方法得到进一步的提高。在氧化剂和催化剂的存在下,由醛与胺合成酰胺化合物被国内外很多课题组所报道 [9] [10] [11] 。由于醛化学性质不稳定,该方法受到了一定的限制。随后汪志勇教授报道了催化氧化醇与胺合成酰胺化合物的方法 [12] ,继汪教授课题组之后,很多课题组也相继报道了催化氧化醇与胺合成酰胺 [13] [14] 。为了开发新的酰胺化合物的合成方法,末端炔烃被作为底物与胺反应合成酰胺 [15] 。虽然这些方法都有效地合成酰胺化合物,但是仍存在反应条件苛刻,底物不稳定,操作复杂等缺点。2014年尹双凤教授课题组报道了铜催化不活泼化合物与三级胺合成酰胺化合物 [16] 。该方法首次通过不活泼C-C键的断裂与三级胺合成酰胺化合物,但是该反应存在反应温度较高,原子经济性不好等缺点。因此,开发一条绿色、经济、环保的酰胺化合物的合成方法是目前的研究热点。本文开发了一种简单、有效、高原子经济性的酰胺化合物的合成方法。该方法以铜为催化剂,取代的苄腈和二级胺为底物,高产率地合成酰胺化合物。这种合成方法具有催化剂廉价,反应条件温和,底物的适用范围广等优点。
2. 实验部分
2.1. 实验仪器与试剂
1H NMR (400 MHz)以TMS为内标,用Bruker 400型核磁共振仪(CDCl3为溶剂);薄层色谱板为青岛海洋化工厂GF254硅胶板;柱层析硅胶为青岛海洋化工厂生产(200~300目);其它化学试剂均为分析纯或化学纯,分别购于安耐吉、百灵威、阿拉丁等试剂厂。
2.2. 实验步骤
铜催化苄腈与二级胺反应合成三级酰胺的一般过程如下:在25 mL的Schlenk管中加入Cu(NO3)2(10 mo%基于底物苄腈),然后抽真空通氧气三次,在氧气氛围下加入0.2 mmol的苄腈化合物,0.24 mmol的二级胺,1 mL甲苯,封管,置于90˚C的油浴锅反应12 h。反应结束后,用饱和NH4Cl水溶液洗涤,乙酸乙酯萃取三次,合并有机溶剂,真空旋干,TLC过柱得到目标产物。
2.3. 目标化合物的结构表征
目标化合物N,N-取代的苯甲酰胺是由铜氧化苄腈,经过C-CN键断裂与二级胺反应合成三级苯甲酰胺。
1) N,N-diethylbenzamide (3a), 1H NMR (400 MHz, CDCl3): δ 7.29 (s, br, 5H), 3.45 (s, br, 2H), 3.16 (s, br, 2H), 1.16 (s, br, 3H), 1.01 (s, br, 3H);
13C
NMR (100 MHz, CDCl3): δ 171.3, 137.2, 129.1, 128.4, 126.2, 43.3, 39.2, 14.2, 12.9.
2) N,N-dipropylbenzamide (3b), 1H NMR (400 MHz, CDCl3): δ 7.36 (s, br, 5H), 3.45 (s, br, 2H), 3.14 (s, br, 2H), 1.67 (s, br, 2H), 1.50 (s, br, 2H), 0.97 (s, br, 3H), 0.72 (s, br, 3H);
13C
NMR (100 MHz, CDCl3): δ 171.8, 137.4, 129.0, 128.4, 126.4, 50.6, 46.2, 21.9, 20.7, 11.4, 11.0.
3) N-cyclohexyl-N-methylbenzamide (3c), 1H NMR (400 MHz, CDCl3): 1H NMR (400 MHz, CDCl3): δ 7.38 (s, br, 5H), 4.52 (s, br, 0.5H), 3.44 (s, br, 0.5H), 2.96 (s, br, 2H), 2.17 (s, br, 2H), 1.04~1.79 (m, 10H);
13C
NMR (100 MHz, CDCl3): δ 171.8, 129.2, 128.5, 126.8, 126.1, 58.3, 52.8, 32.1, 30.8, 29.7, 27.6, 25.6, 25.5, 25.2.
4) phenyl(piperidin-1-yl)methanone (3d), 1H NMR (400 MHz, CDCl3): δ 7.39 (s, br, 5H), 3.71 (s, 2H), 3.34 (s, 2H), 1.67 (s, 4H), 1.51 (s, 2H);
13C
NMR (100 MHz, CDCl3): δ 170.3, 136.5, 129.4, 128.4, 126.8, 48.8, 43.1, 26.6, 25.7, 24.6.
5) morpholino(phenyl)methanone (3e), 1H NMR (400 MHz, CDCl3): δ 7.40 (s, br, 5H), 3.75 (s, br, 4H), 3.62 (s, br, 2H), 3.42 (s, br, 2H);
13C
NMR (100 MHz, CDCl3): δ 170.5, 135.3, 129.9, 128.6, 127.1, 66.9, 66.4, 51.9, 46.2.
6) phenyl(pyrrolidin-1-yl)methanone (3f), 1H NMR (400 MHz, CDCl3): δ 7.51 (s, 2H), 7.39 (s, 2H), 3.64 (s, br, 2H), 3.42 (s, br, 2H), 1.96 (s, br, 2H),1.86 (s, br, 2H);
13C
NMR (100 MHz, CDCl3): δ 169.7, 137.1, 129.8, 128.2, 127.0, 49.6, 46.2, 26.4, 24.4.
7) N,N-diethyl-4-methylbenzamide (3g), 1H NMR (400 MHz, CDCl3): δ 7.27 (d, 2H, J = 7.6 Hz), 7.18 (d, 2H, J = 7.6 Hz), 3.54 (s, 2H), 3.27 (s, 2H), 2.37 (s, 3H), 1.24 (s, br, 3H), 1.11 (s, br, 3H);
13C
NMR (100 MHz, CDCl3): δ 171.5, 139.1, 134.3, 128.9, 126.3, 43.3, 39.3, 21.3, 14.2, 12.9.
8) 4-bromo-N,N-dipropylbenzamide (3h), 1H NMR (400 MHz, CDCl3): δ 7.51 (d, 2H, J = 7.6 Hz), 7.21 (d, 2H, J = 7.6 Hz), 3.42 (s, br, 2H), 3.12 (s, br, 2H), 1.67 (s, br, 2H), 1.51 (s, br, 2H), 0.95 (s, br, 3H), 0.73 (s, br, 3H);
13C
NMR (100 MHz, CDCl3): δ 1701.7, 136.2, 131.6, 128.2, 123.2, 50.7, 49.4, 22.1, 21.9, 11.4, 11.3.
9) N,N-dibenzyl-4-nitrobenzamide (3i), 1H NMR (400 MHz, CDCl3): δ 8.22 (d, 2H, J = 8.0 Hz), 7.63 (d, 2H, J = 8.4 Hz), 7.31~7.38 (m, 8H), 7.11 (d, 2H, J = 6.0 Hz), 4.74 (s, 2H), 4.35 (s, 2H);
13C
NMR (100 MHz, CDCl3): δ 170.1, 148.3, 142.3, 136.3, 135.6, 129.1, 128.9, 128.5, 128.1, 127.9, 127.8, 126.8, 123.9, 51.4, 47.3.
10) N,N-dibenzyl-2-naphthamide (3j), 1H NMR (400 MHz, CDCl3): δ 7.99 (s, 1H), 7.80 (d, 3H, J = 10.0 Hz), 7.58 (d, 1H, J = 8.4 Hz), 7.48 (d, 2H, J = 4.8 Hz), 7.35 (s, 8H), 7.16 ( s, 2H), 4.76 (s, 2H), 4.45 (s, 2H);
13C
NMR (100 MHz, CDCl3): δ 172.4, 137.0, 136.4, 133.7, 133.5, 132.7, 128.9, 128.8, 128.5, 128.4, 127.9, 127.8, 127.7, 127.1, 126.8, 126.6, 124.1, 51.7, 47.0.
3. 结果与讨论
3.1. 反应条件的优化
表1反应条件的优化a。
该反应以苄腈(1a)和二乙胺(2a)作为模板底物,分别考察催化剂、氧化剂、添加剂、溶剂以及温度对反应产率的影响(见表1)。首先以CuCl2为催化剂,氧气为氧化剂,吡啶为添加剂,甲苯为溶剂,90˚C下反应12 h,得到73%的3a (编号1)。接着我们改用不同配体,从结果显示配体的改变并没有提高反应产率(编号2~3)。相反,在不加配体的条件下,3a的产率提高到81% (编号4)。所以我们推断该反应的进行

Table 1. Optimization of the reaction conditionsa
表1. 反应条件的优化a
aReaction conditions: 2-benzyl cyanide (0.2 mmol), diethylamine (0.24 mmol), Cat (0.02 mmol), additive (0.04 mmol), O2 (1 atm), 90˚C, Schlenk for 12 h. bIsolated yield。a反应条件:苄腈(0.2 mmol),二乙胺(0.24 mmol),催化剂(0.02 mmol),添加剂(0.04 mmol), O2 (1 atm),90˚C,Schlenk中反应12 h。b分离产率。
不需要额外增加配体,所以,以甲苯为溶剂,在不添加配体的条件下,我们尝试不同铜催化剂对该反应的影响(编号5~9),当以Cu(OAc)2、Cu(NO3)2、CuBr2、CuBr、CuI为催化剂时,Cu(NO3)2显示了很好的催化效果,3a产率高达90% (编号6)。接着以Cu(NO3)2为催化剂,尝试不同溶剂对反应的的影响(编号10~12),结果显示该反应在DMF、CH3CN中产率较低,在1,4-二氧六环中收率中等,从以上结果显示,甲苯是该反应的最优溶剂。最终我们确定该反应的最优条件为:催化剂为Cu(NO3)2,(0.02 mmol),溶剂为甲苯(1 mL),无添加剂,温度为90˚C,反应时间12 h。
3.2. 反应底物的拓展
在最优反应条件下,考察了底物腈和胺的适用范围(见图1)。首先考虑不同的二级胺对反应的影响,除二乙胺外,二正丙胺为胺底物与苄腈反应,酰胺3b的收率为86% (编号2)。结果显示该反应与胺的链长没有关系。当以N-甲基环己胺为底物,N-甲基-N-环己基苯甲酰胺3c的收率为85%,该反应结果证明即使胺分子连有位阻较大的环己基,也不会阻碍反应的发生(编号3)。当以哌啶、吗啡啉、吡咯烷为底物与苄腈反应,相应酰胺的收率都较高(编号4~6)。从胺反应的结果可以看出,无论是链状胺还是环状胺都可以作为很好的底物,与苄腈反应得到相应的酰胺。随后我们考虑苄腈芳环上取代基对反应的影响,当苄腈芳环连有给电子取代基甲基1b,与胺2a反应也可以得到相应的酰胺3g,产率83% (编号7)。当芳环上连有卤素溴取代基1c,也可以与胺2b反应得到相应的酰胺3h,收率为86% (编号8)。当强吸电子基团硝基存在时,比如1d作为底物可以与2-苄胺2 g反应生成相应的酰胺3i,产率86% (编号9)。从结果可以看出,无论苄腈芳环上连有给电子取代基还是吸电子取代基,都可以与二级胺反应得到相应的酰胺产物。当以2-萘乙腈1e为反应底物时,也可以与胺2g反应得到相应的酰胺3j (标号10)。
图1反应底物的拓展a。
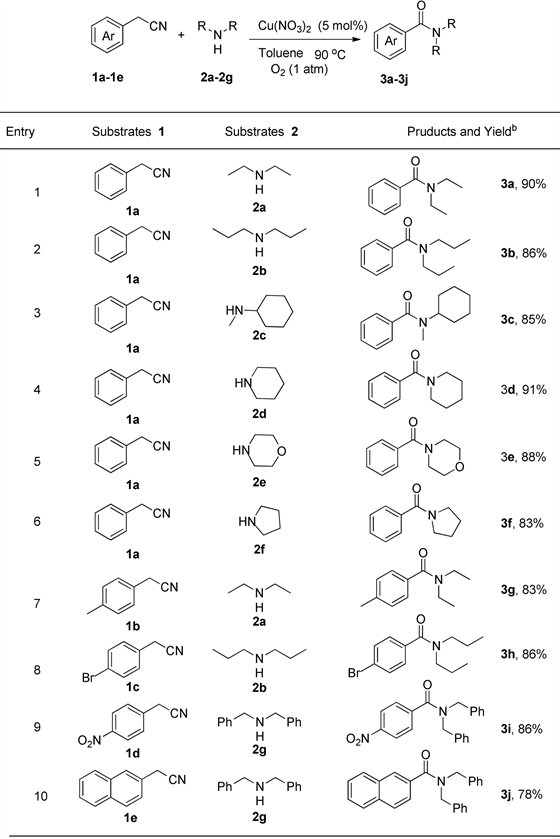 aReaction conditions: substituted-benzyl cyanide (0.2 mmol), secondary amine (0.24 mmol), Cu(NO3)2 (0.02 mmol, 10%), O2 (1 atm), 90˚C, Schlenk for 12 h. bIsolated yield。a反应条件:取代的芳腈(0.2 mmol),二级胺(0.24 mmol),Cu(NO3)2 (0.02 mmol, 10%),O2 (1 atm),90˚C,Schlenk中反应12 h。b分离产率。
aReaction conditions: substituted-benzyl cyanide (0.2 mmol), secondary amine (0.24 mmol), Cu(NO3)2 (0.02 mmol, 10%), O2 (1 atm), 90˚C, Schlenk for 12 h. bIsolated yield。a反应条件:取代的芳腈(0.2 mmol),二级胺(0.24 mmol),Cu(NO3)2 (0.02 mmol, 10%),O2 (1 atm),90˚C,Schlenk中反应12 h。b分离产率。
Figure 1. Expansion of reactive substratea
图1. 反应底物的拓展a
3.3. 可能的反应机理
为了探索该反应可能的反应历程,我们做了相应的控制实验(见图2),首先以苯甲醛1f为反应底物,在标准条件下与二级胺反应,结果没有得到想要的酰胺(见图2 Eq.1),此结果说明,该反应不是苄腈变为醛再与胺反应生成酰胺产物。接着以α-羰基苯乙腈为底物,与二级胺反应,结果显示在较短的反应内,所要酰胺的产物高达91%,从此结果可以看出,在苄腈反应过程中,苄腈首先氧化为α-羰基苯乙腈化合物再与胺反应生成相应的酰胺产物(见图2 Eq.1)。
根据以上控制实验结果和文献报道 [17] ,铜催化苄腈与二级胺反应生成酰胺的反应机理可能如下(见图3):首先二级胺2在铜离子和氧气的作用下生成中间体铜胺离子I2a,苄腈1被氧化为相应的α-羰基腈I1a,然后中间体胺离子I2a进攻α-羰基腈I1a生成相应的酰胺3,释放出铜离子进行下一步的催化反应。
4. 结论
本文开发了一种简单有效的铜催化苄腈与二级胺反应合成三级酰胺化合物的新体系,并且提出了可能的反应机理。该方法一步实现了C-H键的氧化,C-C键的断链和C-N键的形成。该反应体系的提出为酰胺化合物的合成开辟了一种新的合成途径。该反应体系下,产物的选择性和产率都很高,催化剂廉价,底物适用范围广,是一种酰胺化合物的绿色合成方法。
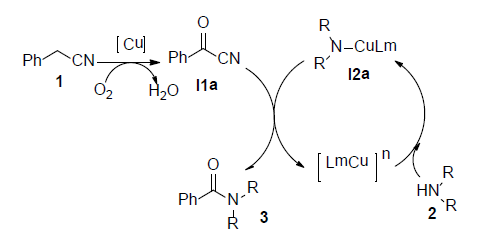
Figure 3. Reaction Mechanism by Copper Catalyzed
图3. 铜催化苄腈与二级胺反应合成三级酰胺的反应机理
致谢
感谢国家自然科学基金(No. 21603068),和湖北科技学院博士启动基金(2016-19XB011)对本工作的支持与帮助。
[1] 盛国柱, 张炜. 酰胺官能团构建方法研究新进展[J]. 有机化学, 2013(33): 2271-2282.
[2] Shen, B., Makley, D.M. and Johnston, J.N. (2010) Umpolung Reactivity in Amide and Peptide Synthesis. Nature, 465, 1027-1033. https://doi.org/10.1038/nature09125
[3] Valeur, E. and Bradley, M. (2009) Amide Bond Formation: Beyond the Myth of Coupling Reagents. Chemical Society Reviews, 38, 606-631. https://doi.org/10.1039/B701677H
[4] Humphrey, J.M. and Chamberlin, A.R. (1997) Chemical Synthesis of Natural Product Peptides: Coupling Methods for the Incorporation of Noncoded Amino Acids into Peptides. Cheminform, 97, 2243-2266.
[5] Castanho, M. and Santos, N. (2011) Peptide Drug Discovery and Development. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. https://doi.org/10.1002/9783527636730
[6] Li, M., Hu, L., Cao, X.Q., et al. (2011) Direct Hydrogenation of Nitroaromatics and One-Pot Amidation with Carboxylic Acids over Platinum Nanowires. Chemistry, 17, 2763-2768. https://doi.org/10.1002/chem.201002801
[7] Ghaffarzadeh, M., Joghan, S.S. and Faraji, F. (2012) A New Method for the Synthesis of Amides from Imines. Tetrahedron Letters, 53, 203-206. https://doi.org/10.1016/j.tetlet.2011.11.018
[8] Starkov, P. and Sheppard, T.D. (2011) Borate Esters as Convenient Reagents for Direct Amidation of Carboxylic Acids and Transamidation of Primary Amides. Organic & Biomolecular Chemistry, 9, 1320-1323. https://doi.org/10.1039/c0ob01069c
[9] Xie, S., Zhang, Y., Ramström, O., et al. (2016) Base-Catalyzed Synthesis of Aryl Amides from Aryl Azides and Aldehydes. Chemical Science, 7, 713-718. https://doi.org/10.1039/C5SC03510D
[10] Liu, Z.J., Zhang, J. and Chen, S.L. (2012) Cross Coupling of Acyl and Aminyl Radicals: Direct Synthesis of Amides Catalyzed by Bu4NI with TBHP as an Oxidant. Angewandte Chemie International Edition, 51, 3231-3235. https://doi.org/10.1002/anie.201108763
[11] Kekeli, E.K. and Wolf, C. (2007) Metal-Free One-Pot Oxidative Amination of Aldehydes to Amides. Organic Letters, 9, 3429-3232. https://doi.org/10.1021/ol7014626
[12] Xu, K., Hu, Y.B., Wang, Z.Y., et al. (2012) Direct Amidation of Alcohols with N-Substituted Formamides under Transition-Metal-Free Conditions. Chemistry-A European Journal, 18, 9793-9797. https://doi.org/10.1002/chem.201201203
[13] Xiao, F., Liu, Y., Deng, G., et al. (2012) Peroxide-Mediated Transition-Metal-Free Direct Amidation of Alcohols with Ni troarenes. Organic Letters, 14, 984-987. https://doi.org/10.1021/ol203211k
[14] Ghosh, S.C., Muthaiah, S., Zhang, Y., et al. (2009) Direct Amide Synthesis from Alcohols and Amines by Phosphine-Free Ruthenium Catalyst Systems. Advanced Synthesis & Catalysis, 351, 2643-2649. https://doi.org/10.1002/adsc.200900482
[15] Wei, W., Hu, X.Y., Yan, X.W., et al. (2012) Direct Use of Dioxygen as an Oxygen Source: Catalytic Oxidative Synthesis of Amides. Chemical Communications, 48, 305-307. https://doi.org/10.1039/C1CC14640H
[16] Chen, X., Zhou, Y., Yin, S., et al. (2014) Copper-Catalyzed Aerobic Oxidative Inert C-C and C-N Bond Cleavage: A New Strategy for the Synthesis of Tertiary Amides. Chemistry-A European Journal, 20, 1-6. https://doi.org/10.1002/chem.201403144
[17] Guo, S., Qian, B., Xie, Y., et al. (2011) Copper-Catalyzed Oxidative Amination of Benzoxazoles via C-H and C-N Bond Activation: A New Strategy for Using Tertiary Amines as Nitrogen Group Sources. Organic Letters, 13, 522-525. https://doi.org/10.1021/ol1030298
Supporting Informations
https://image.hanspub.org/pdf/3160051_paper_images.pdf