摘要: 目的:鉴定对结肠腺癌患者具有潜在预后价值的免疫相关长链非编码RNA。方法:从癌症基因组图谱(The Cancer Genome Atlas, TCGA)数据库中获得结肠腺癌样本的临床信息和全基因表达数据,根据分子特征数据库,找到免疫相关的差异基因。使用相关性分析,找到免疫相关的差异表达长链非编码RNA (lncRNA),最后依据病人的生存状态,通过cox回归分析找出7个免疫和生存相关的差异表达lncRNA。依据这7个lncRNA在病人中的表达量,构建Cox风险模型,计算病人的风险值,通过中位数,将病人分为高低风险组。使用临床特征进行单因素和多因素独立预后分析,查看临床特征与预后之间的吻合概率。通过相关性分析检测在不同临床特征中7个lncRNA的表达情况。通过受试者工作特征曲线(receiver operator characteristic curve, ROC)检测预后因素的准确性。依据主成分分析(Principal Component Analysis, PCA),观察各样品的基因表达模式。最后通过基因富集分析(Gene Set Enrichment Analysis, GSEA)和免疫相关性分析,观察基因分布情况。结果:本研究共收录437个队列研究,其中癌组织为398个,正常组织为39个。通过相关性分析找到20个免疫相关的差异表达lncRNA。确定了7个最具预后价值的免疫相关lncRNA (AC245100.7, AP001189.3, LINC01503, ZEB1-AS1, AC004585.1, SNHG16, AP006621.3)。构建Cox风险模型,发现高风险组的病人的死亡率显著增高,五年生存率显著降低。通过单因素和多因素独立预后分析,发现年龄,临床分期以及风险值可以作为独立预后因子。相关性分析表明ZEB1-AS1在N分期中表达与疾病分期高度一致。ROC曲线显示临床分期,N分期和风险值均有较好的临床预后。PCA分析发现基7个预后相关的lncRNA可以很好的将高低风险组区分开。通过GSEA富集分析,发现免疫反应和免疫进程的相关基因在高风险的病人体内明显富集。通过免疫相关性分析发现高风险组患者中B细胞、T细胞和巨噬细胞相关基因表达升高以及副炎症、I型干扰素和II型干扰素相关基因表达显著升高。结论:7个免疫相关的预后lncRNA对结肠腺癌具有预后价值。
Abstract:
Objective: Identification of immune-related long-chain non-coding RNAs with potential prognostic value for colon adenocarcinoma patients. Methods: Obtain clinical information and full gene expression data of colon adenocarcinoma samples from the Cancer Genome Atlas (TCGA) database, and find immune-related differential genes according to the molecular feature database. Use correlation analysis to find immune-related differentially expressed long non-coding RNA (lncRNA). Finally, according to the patient’s survival status, seven immune-related and survival-related differentially expressed lncRNAs were found through cox regression analysis. Based on the expression levels of these 7 lncRNAs in patients, a Cox risk model was constructed, the patient’s risk value was calculated, and the patients were divided into high and low risk groups based on the median. Use clinical features to perform univariate and multivariate independent prognostic analysis to view the probability of agreement between clinical features and prognosis. Correlation analysis was used to detect the expression of 7 lncRNAs in different clinical features. The accuracy of prognostic factors is detected by receiver operator characteristic curve (ROC). According to principal component analysis (Principal Component Analysis, PCA), observe the gene expression pattern of each sample. Finally, through Gene Set Enrichment Analysis (GSEA) and immune correlation analysis, observe the distribution of genes. Results: This study included a total of 437 cohort studies, including 398 cancer tissues and 39 normal tissues. Through correlation analysis, 20 immune-related differentially expressed lncRNAs were found. Seven immune-related lncRNAs with the most prognostic value were identified (AC245100.7, AP001189.3, LINC01503, ZEB1-AS1, AC004585.1, SNHG16, AP006621.3). The Cox risk model was constructed and it was found that the mortality rate of patients in the high-risk group was significantly increased, and the five-year survival rate was significantly reduced. Through univariate and multivariate independent prognostic analysis, it is found that age, clinical stage and risk value can be used as independent prognostic factors. Correlation analysis showed that the expression of ZEB1-AS1 in the N stage was highly consistent with the disease stage. ROC curve shows clinical stage, N stage and risk value have good clinical prognosis. PCA analysis found that based on 7 prognostic-related lncRNAs, high-risk groups can be well distinguished from low-risk groups. Through GSEA enrichment analysis, it is found that genes related to immune response and immune process are significantly enriched in high-risk patients. Through immune correlation analysis, it was found that the expression of B cells, T cells, and macrophages related genes in the high-risk group was increased, and the expression of para- inflammation, type I interferon and type II interferon related genes were significantly increased. Conclusion: Seven immune-related prognostic lncRNAs have prognostic value for colon adenocarcinoma.
1. 引言
结肠腺癌是消化系统最常见的恶性肿瘤之一。其发病率随着生活水平的提高呈增长的趋势。其在中国癌症发病率居第三位,死亡率居第五位 [1]。控制发病率及降低死亡率是结肠腺癌防治工作的重点,而根据预后预测指导结肠腺癌的治疗是降低死亡率的一种有效手段。因此,迫切需要寻找一种敏感性及特异性高的指标,预测结肠腺癌的发生、发展,制定个体化治疗方案。
长链非编码RNA是一类细胞内源性的RNA,一般长度大于200个核苷酸,缺乏蛋白质编码能力 [2]。研究表明,长链非编码RNA参与结肠腺癌的发生发展,起到重要的调控作用,包括癌症的发生、发展和预后 [3]。如SNHG6 and CTD-2354A18.1可以作为独立预后因子可以预测结肠腺癌的预后 [4]。
免疫系统可以影响癌症进展的观点一直是研究热点。研究表明肿瘤免疫微环境在预测预后和评估治疗功效因子方面具有重要价值 [5]。肿瘤微环境由免疫细胞,免疫相关途径和免疫细胞分泌的细胞因子组成。在结肠癌中,已有研究表明,适应性免疫反应与生存结果和复发密切相关 [6]。
生物信息学的发展极大促进结肠腺癌的机制以及预后标志物的研究,发现HOTAIR,LINC00355,KCNQ1OT1和TSSC1-IT1与总生存率呈负相关 [7]。但是在结肠腺癌中免疫相关的lncRNA还缺乏深入的研究。我们通过整合挖掘基因表达数据库数据,并进行全面的生物信息学分析,筛选出了具有预后作用的免疫相关lncRNA。为结肠腺癌的治疗提供了新的标志物,也为结肠腺癌发生发展机制提供了新思路。
2. 材料和方法
2.1. 样本数据的收集
通过TCGA数据库,提取结肠腺癌的表达数据和临床性状数据,本研究共收录437份结肠腺癌样本。在进行Cox生存分析是生存期 ≤ 10天以及资料不全的样本剔除。
2.2. 免疫及预后相关的差异表达lncRNA
免疫信号通路以及免疫反应分子数据集是从分子特征数据库v4.0中获得的(免疫系统过程M13664,免疫反应M19817,(https://www.gsea-msigdb.org/gsea/msigdb/index.jsp)。lncRNA基因集来自于GENCODE (http://www.gencodegenes.org)。通过统计免疫相关的差异基因以及lncRNA,进行相关性分析,找到免疫相关的差异lncRNA。再将得到的免疫相关的差异表达基因与临床预后进行Cox回归分析,得到免疫及预后相关的差异表达lncRNA。
2.3. 预后相关lncRNA风险评估以及风险模型的建立
通过Cox风险回归模型,计算预后相关lncRNA的风险系数,再使用预后相关lncRNA在每个病人的表达量乘以风险系数,所有的预后相关lncRNA的累计值为每个病人的风险值。具体公式为:风险评分 = lncRNA1表达量 × 风险回归系数1 + lncRNA2表达量 × 风险回归系数2 + … + lncRNA7表达量 × 风险回归系数7。
2.4. 生物信息学分析
通过Cox风险回归模型,计算病人的风险值,以中位值为界将病人分为高低风险组,观察高低风险组的生存状态并绘制生存曲线。通过单因素和多因素独立预后分析检测临床性状以及风险值作为预后因子的可能性。通过ROC曲线检测预后因子的准确性。通过相关性分析,检测预后lncRNA在不同临床性状中的表达情况。通过主成分分析检测基因在高低风险组中的表达情况。通过GSEA富集分析,检测高低风险组基因富集情况。通过免疫相关性分析,评估免疫细胞和免疫功能相关基因富集情况。
2.5. 统计分析
应用Cox回归分析,构建风险模型。通过单因素和多因素独立预后分析,统计病人临床性状与预后之间的关系,ROC曲线分析预后因子的准确性,相关性分析预后lncRNA在不同临床分期中的表达情况,使用PCA和GSEA分析基因富集情况。所有统计分析均使用R语言4.0.2版本。P值 < 0.05被认为是有显著意义。
3. 结果
3.1. 获得预后及免疫相关的lncRNA及对模型的评估
从TCGA平台上下载了437个食管鳞状细胞癌基因组样本。同时从分子特征数据库中收集了332个免疫相关基因。通过筛选得到免疫相关的差异lncRNA,再依据临床性状通过Cox 比例风险回归模型,得到20条预后及免疫相关的差异lncRNA (图1(A))。根据病人的风险值,区分高低风险组(图1(B)),观察高低风险组病人的生存状态,发现高风险组的病人死亡人数和死亡率均有明显的升高(图1(C))。通过绘制风险热图,发现AC245100.7,AP001189.3,LINC01503,ZEB1-AS1,AC004585.1和AP006621.3随着风险值的升高而升高,为高风险的基因,而SNHG16随着风险值的升高而降低,为低风险基因(图1(D))。通过模型将计算病人的风险值,以中位数为临界点将病人区分为高风险和低风险两组,通过R语言绘制生存分析曲线,发现两组有显著差异(图1(E)),说明我们的模型可以很好的预测病人的预后。
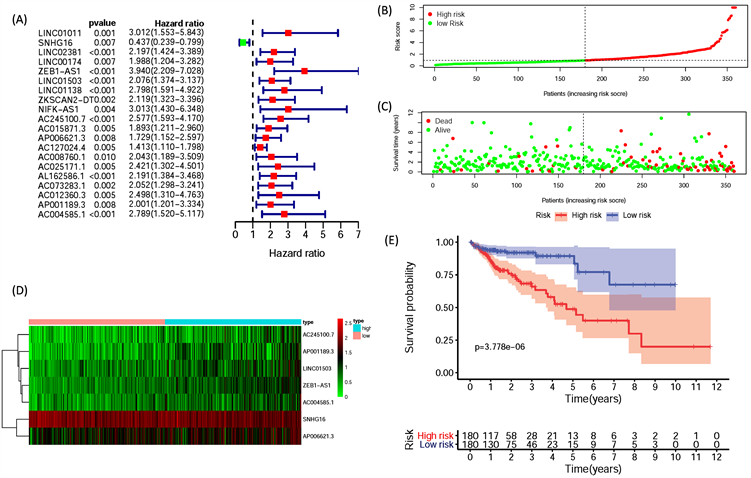 (A) 病人分为高风险组和低风险组。(B) 高风险组和低风险组病人生存状态。(C) 六条lncRNA在高低风险组中的表达情况。(D) 高低风险组的生存预后。
(A) 病人分为高风险组和低风险组。(B) 高风险组和低风险组病人生存状态。(C) 六条lncRNA在高低风险组中的表达情况。(D) 高低风险组的生存预后。
Figure 1. Construct a risk assessment model
图1. 构建风险评估模型
3.2. 临床病理特征分析以及风险模型评估
通过单因素独立预后分析,发现分期(stage)、肿瘤浸润深度(T)、是否区域淋巴结转移(N)以及风险值有显著性,而年龄和性别没有差异(图2(A)),进一步通过多因素独立预后分析,发现年龄、分期(stage)以及风险值均有显著差异(图2(B)),说明以上临床性状可以有效的预测病人预后。为了进一步验证预后及免疫相关的lncRNA与临床分期相关性,以性别、分期(stage)、是否区域淋巴结转移(N)、肿瘤浸润深度(T)、是否远处转移(M)以及风险值分组,计算风险lncRNA的表达情况,发现在临床分期、T分期、N分期中ZEB1-AS1随着病情的发展表达逐渐升高,说明ZEB1-AS1是一个与多个临床分期高度相关的lncRNA (图2(C)~(E))。通过ROC曲线比较病人五年生存率,计算曲线下面积评估评价指标对疾病预后的准确性,发现风险模型预测结果较好,曲线下面积(Area Under Curve, AUC)为0.694 (图2(F))。说明构建的风险评估模型可以较好预测病人的预后情况。
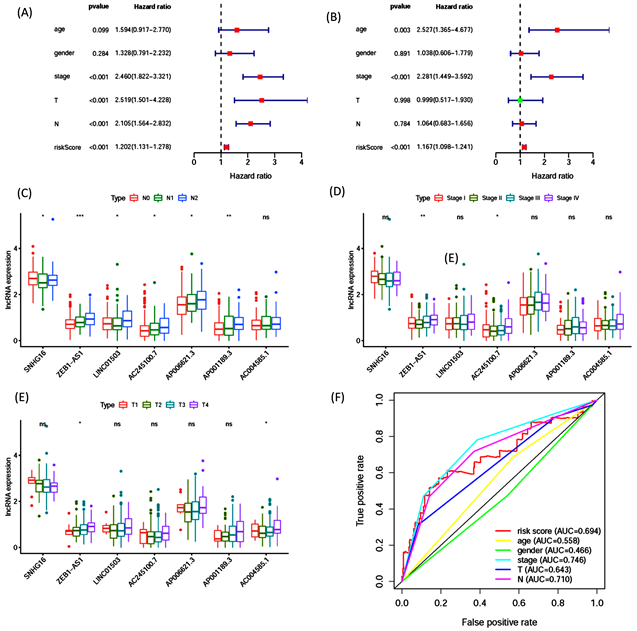 (A) 单因素独立预后分析评估年龄、性别、分期(stage)、是否区域淋巴结转移(N)、肿瘤浸润深度(T)、是否远处转移(M)以及风险值对病人预后的评估。(B) 多因素独立预后分析评估年龄、性别、分期(stage)、是否区域淋巴结转移(N)、肿瘤浸润深度(T)、是否远处转移(M)以及风险值对病人预后的评估。(C) 检测7条预后相关风险lncRNA在N分期中的表达情况。(D) 检测7条预后相关风险lncRNA在临床分期中的表达情况。(E) 检测7条预后相关风险lncRNA在T分期中的表达情况。(F) 评估临床分期、性别、年龄、T分期、N分期和风险模型对病人预后预测的准确性。
(A) 单因素独立预后分析评估年龄、性别、分期(stage)、是否区域淋巴结转移(N)、肿瘤浸润深度(T)、是否远处转移(M)以及风险值对病人预后的评估。(B) 多因素独立预后分析评估年龄、性别、分期(stage)、是否区域淋巴结转移(N)、肿瘤浸润深度(T)、是否远处转移(M)以及风险值对病人预后的评估。(C) 检测7条预后相关风险lncRNA在N分期中的表达情况。(D) 检测7条预后相关风险lncRNA在临床分期中的表达情况。(E) 检测7条预后相关风险lncRNA在T分期中的表达情况。(F) 评估临床分期、性别、年龄、T分期、N分期和风险模型对病人预后预测的准确性。
Figure 2. Clinic pathological characteristics analysis and risk model evaluation
图2. 临床病理特征分析及风险模型评估
3.3. 高低风险组在不同基因分组中的主成分差异
为了进一步明确在不同基因分组中,基因分布情况,通过主成分分析的方法将基因的分布进行可视化,发现在风险模型中,高低风险组可以很好的区别开来,而在全基因组、免疫基因和免疫lncRNA中基因无法完全区别开来(图3(A)~(D))。
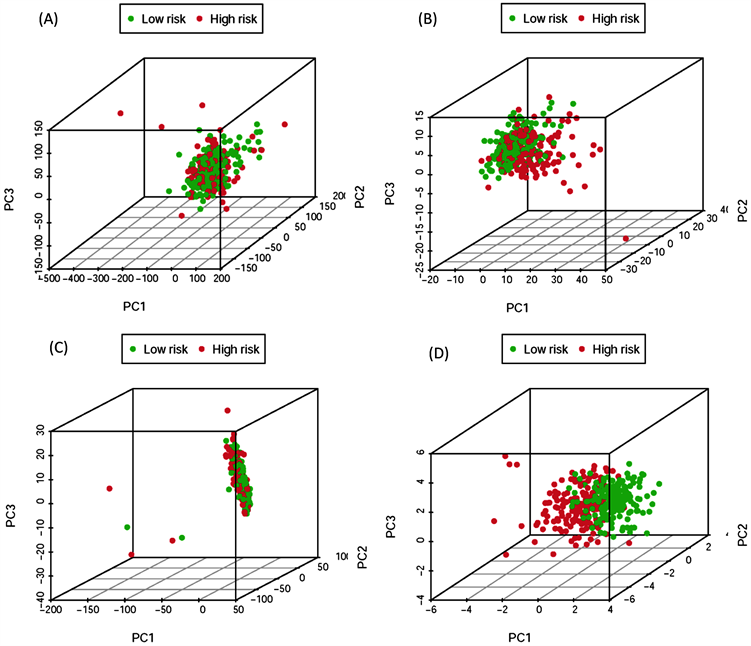 (A) 全基因在高低风险人群中的分布情况。(B) 免疫基因在高低风险人群中的分布情况。(C) 免疫相关lncRNA基因在高低风险人群中的分布情况。(D) 风险基因在高低风险人群中的分布情况。
(A) 全基因在高低风险人群中的分布情况。(B) 免疫基因在高低风险人群中的分布情况。(C) 免疫相关lncRNA基因在高低风险人群中的分布情况。(D) 风险基因在高低风险人群中的分布情况。
Figure 3. Principal component analysis of various genetic data of high- and low-risk groups
图3. 高低风险人群各类基因数据的主成分分析
3.4. 免疫相关基因分析
通过GSEA富集分析,发现在高低风险组的病人中,免疫反应相关基因以及免疫系统进程相关基因均在高风险病人中富集,说明与免疫相关的分子以及信号通路在风险模型中存在显著差异(图4(A),图4(B))。通过免疫相关性分析,在免疫相关的细胞基因分析中发现在高风险组患者中B细胞、T细胞和巨噬细胞相关基因表达升高(图4(C)),而在免疫相关功能分析中发现高风险组患者中副炎症、I型干扰素和II型干扰素相关基因表达显著升高(图4(D))。
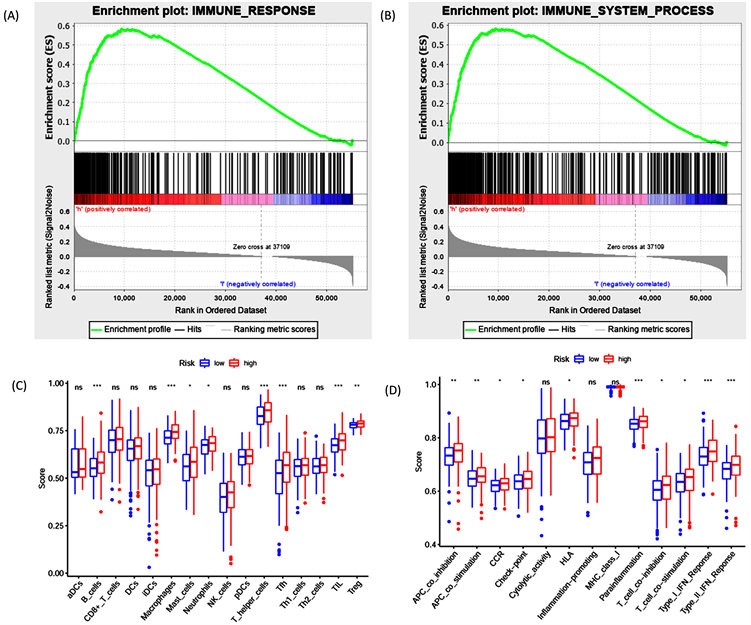 (A) 高低风险组中免疫反应相关分子的富集情况。(B) 高低风险组中免疫系统进程相关分子的富集情况。(C) 免疫细胞相关性分析。(D) 免疫功能相关性分析。
(A) 高低风险组中免疫反应相关分子的富集情况。(B) 高低风险组中免疫系统进程相关分子的富集情况。(C) 免疫细胞相关性分析。(D) 免疫功能相关性分析。
Figure 4. GSEA enrichment analysis and immune correlation analysis
图4. GSEA富集分析和免疫相关性分析
4. 讨论
随着高通量测序技术的快速发展和相关技术的广泛应用,生物医学研究已进入后基因组时代,大规模组学数据呈指数级增长。目前,生物计算和生物信息学可以帮助从复杂数据中挖掘出有意义且规则的基因。为了找出相关的生物过程的重要分子机制,研究者通常在各种统计和计算模式的基础上寻找差异表达的基因,然后对差异表达的基因进行功能富集分析,以揭示和理解基本的分子机制 [8]。
结肠癌是一种多因素疾病,其病因包括遗传因素,环境暴露和消化道的炎症。尽管传统的大肠癌预后和预测因素,例如年龄,肿瘤分期,手术切缘,受影响的局部淋巴结数目和肿瘤等级,已在患者临床预后方面取得了显著效果,但它们在区分相关癌症风险亚组时显示出明显的局限性。由于分子异质性而具有不同的临床结果 [9]。因此,在过去的十年中,已经在广泛的临床转录组研究中系统地研究了分子标志物的预后潜力 [10]。
在我们的研究中,我们最终筛选了20个差异的免疫相关lncRNA,进一步通过将lncRNA与风险值进行Cox分析,得出7个免疫相关风险lncRNA,通过五年生存率的比较,发现高低风险组的生存率有显著差异,说明风险模型可以很好的区分病人生存率。通过单因素独立预后分析和多因素独立预后分析比较其年龄、性别、临床分期以及T、N分期和风险值,发现年龄、临床分期以及风险值可以作为结肠腺癌的独立预后因子。进一步分析各临床特征中不同的风险lncRNA的表达情况,发现ZEB1-AS1的表达在N分期、T分期和临床分期中表达随着病情恶化表达也增高,说明ZEB1-AS1可能在结肠腺癌的发生、发展中起重要作用。最后通过ROC曲线验证预后模型的准确性,发现风险值、临床分期以及N分期均能较好的预测病人预后。进一步通过PCA主成分分析,发现在高低风险组中风险lncRNA的表达区分最为明显。最后,统计高低风险组中免疫相关基因的GSEA富集分析,发现不管在免疫进程还是免疫反应相关基因均有差异,说明免疫基因在高低风险组中有显著差异,在疾病发生发展过程中起重要作用。
总而言之,我们的研究确认了7个免疫相关风险lncRNA作为结肠腺癌患者独立预后因子,同时为结肠腺癌免疫学机制提出了新的分子基础。这些结果为我们进一步研究免疫相关lncRNA在结肠腺癌中的发病机制以及临床预后的预测提供了思路。
基金项目
褪黑素受体2在阿尔兹海默病中的作用及机制(B2018261)。
参考文献