1. 引言
来源于植物、动物、微生物等的天然产物是药物开发的重要源泉 [1] [2] [3] [4] [5]。据统计,1981~2019全世界所批准上市的1881个小分子实体药物中,天然源药物占比约达46% [6]。其中,在自然界广泛存在的微生物及其次生代谢产物作为新药发现与创制的资源宝库,引起药物研究者的关注。微生物药物具有结构和作用机制的多样性 [7] [8] [9],以及良好的生物安全性和相容性,药物化学家们开发出了青霉素、头孢菌素等中药的抗生素类药物;此外,农药学家们开发出了枯草芽孢杆菌和咯菌腈等微生物药物 [8] [10],为人类的生命安全和作物健康提供了有力保障。在微生物药物中,抗生素 [7] 是一类具有抗细菌感染、免疫调节功能和抗肿瘤活性的化合物。早在1928年细菌学家与生物学家弗莱明首次从青霉菌的原液中发现了青霉素,由此开启了抗生素研究的历史新篇章,诞生了如庆大霉素、林可霉素、螺旋霉素、链霉菌素、链霉素、罗红霉素和亚胺培南以及卡那霉素等系列抗生素。
然而,由于抗生素的滥用,日益增长的细菌耐药问题成为直接威胁人类健康与全球经济良性发展的原因之一 [11]。随着多重耐药的革兰氏阳性和革兰氏阴性菌的不断出现,大多数市售抗生素的临床疗效被广泛限制。因此,开发新型高效抗生素具有极其重要的科学意义与研究价值。吡咯霉素是一类含有广泛生物活性的微生物代谢产物,如抗细菌、抗真菌、驱虫、抗细胞增殖、杀虫和除螨等 [12],在药物的开发与应用方面具有广阔的前景。本文综述了吡咯霉素类化合物的发现及结构、生源合成、全合成及其生物活性研究进展,以期为后续研究奠定基础。
2. 天然吡咯霉素的发现及结构
天然吡咯霉素(pyrrolomycins)是一类具有较强的抑菌活性的多卤代抗生素类化合物,其来源是放线菌属和链霉菌属等的次生代谢产物 [13],该类化合物的结构特征为 [14]:1) 高卤化度,多为氯取代;2) 吡咯环的3位硝基增加了部分化合物的活性。天然吡咯霉素的抗菌活性可能与卤素含量相关,其随卤化程度越高,活性显著增强 [15] [16]。
二十世纪六十年代中期,Arima等 [17] 首次从吡咯菌素假单胞菌(Pseudomonas pyrrocinia)中发现了首个吡咯类抗生素——硝吡咯菌素,并经衍生化得到了芳基吡咯类拌种咯和咯菌腈。1981年Kaneda等 [18] [19] 在筛选新型抗菌剂的过程中,从未经处理的放线菌属(Streptomyces) SF-2080培养液中发现了两种特殊物质,经结构确认为pyrrolomycin A (1)、pyrrolomycin B (2)。1983年Ezaki等 [20] [21] 在放线菌(Actinosporangiuml ritaminophilian) SF-2080培养液中发现了pyrrolomycin C (3)、D (4)和E (5)。同年他们 [22] 从同种菌的不同培养液中发现与3的结构相似但含有溴原子的化合物 pyrrolomycin F1 (6)、F2a (7)、F2b (8)和F3 (9)。2004年Charan等 [23] 为探究生物系统硝化机制的原理,用富马链球菌(Streptomyces fumanus)培养基LL-F42248进行发酵实验,5天后得到dioxapryrrolomycin (10),培养基经二次发酵得到pyrrolomycin G-J (11-14)和pyrrolomycin C (3),在这些吡咯霉素家族成员中,只有dioxapryrrolomycin (10)、pyrrolomycin G (11)和pyrrolomycin H (12)具有手性中心(如图1所示)。

Figure 1. Natural pyrrolomycins compounds
图1. 天然吡咯霉素类化合物
3. 吡咯霉素生源合成
由于吡咯霉素的骨架结构特征,其硝基的来源一直是合成生物学家们关注的重点,为探索吡咯霉素的硝化机制,生物化学家们开展了吡咯霉素生物合成途径的研究,提出了生源合成假说。
3.1. Carter的吡咯霉素生物合成
2006年,Carter等 [24] 为探索dioxapryrrolomycin (10)的合成与次生代谢产物pyrrolomycin G-H (11-12)之间的生物转化关系,开展了链霉菌(Streptomyces fumanus)中的dioxapryrrolomycin (10)的生物合成研究工作。如图2所示,采用同位素喂养法,以L-脯氨酸和乙酸作为起始原料,经过系列生物合成转化得到吡咯霉素骨架,进一步转化得到pyrrolomycin C (3),再经过硝化、羰基不对称还原及芳构化得到pyrrolomycinG (11),用13C标记的S-腺苷甲硫氨酸进行O-甲基化得到pyrrolomycin H (12),进一步氧化环化得到dioxapryrrolomycin (10)。pyrrolomycin C (3)是其他吡咯霉素的生物合成前体,同时发现细菌培养物LL-F42248利用大气N2O为氮源实现酶催化硝化。综合研究表明天然吡咯霉素结构中吡咯环和桥碳C-6来源于L-脯氨酸,苯基部分来源于三个乙酸酯单元的缩合,硝基来源于大气中的N2O,苯酚甲醚中甲基来源于S-腺苷甲硫氨酸。
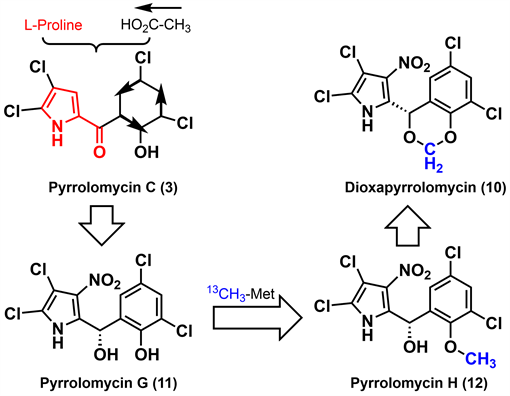
Figure 2. Carter’s biosynthesis of pyrrolomycins [24]
图2. Carter等的吡咯霉素生源合成 [24]
3.2. Parry的吡咯霉素生源合成假说
2007年,Parry等 [13] 探究了吡咯霉素生物合成中的硝化机制,通过维生素放线菌(Actinosporangium vitaminophilum)吡咯霉素生物合成基因簇的克隆技术,提出了一种吡咯霉素类化合物生物合成途径。如图3所示,吡咯霉素基因簇中编码的硝酸还原酶可能在产生活性亚硝基化物中起着重要作用,但没有揭示将硝基引入这些抗生素的确切机制。基于可获得的信息,Parry等推测相互转化过程的机理有三种可能性。第一种是亲核芳香取代反应,pyrrolomycin C (3)氯化为pyrrolomycin D (4),4与亚硝酸盐反应生成2,3-二氯-4-硝基吡咯,该吡咯可转化为pyrrolomycin A (1)和pyrrolomycin B (2)以及dioxapryrrolomycin (10)。第二种是亚硝酸盐与pyrrolomycin C (3)的C-4亲核加成后,生成2,3-二氯-4-硝基吡咯,再经系列反应生成不同的吡咯霉素。第三种是亲电芳香取代反应,pyrrolomycin C (3)的衍生物与亚硝酸产生的亚硝基阴离子发生反应,生成2,3-二氯-4-亚硝基吡咯,然后将其氧化成相应的硝基吡咯。尽管最近研究的吡咯霉素生物合成方面取得了一定进展,但生物化学硝化的细节仍不清楚。
4. 吡咯霉素化学合成及生理活性研究
多种微生物中产生一种具有广泛生物活性的吡咯霉素化合物,其具有不同程度的抗菌、杀虫和杀螨活性、细胞毒活性 [13]。但由于其存在生物分离效率低、来源受到限制的问题,自1981年Koyama等报道pyrrolomycin A (1)和pyrrolomycin B (2)的结构、合成及生物活性研究以来,越来越多的科学家投入到吡咯霉素化合物的化学合成及生理活性研究中。
4.1. 天然吡咯霉素类化合物
天然吡咯霉素是一类由链霉菌属和放线菌属产生的多卤代代谢产物,其对革兰氏阳性菌和革兰氏阴性菌均具有良好的抑制效果。但截至目前为止,关于天然吡咯霉素的合成报道较少,且多数存在收率低、副产物多、反应选择性较差的问题。
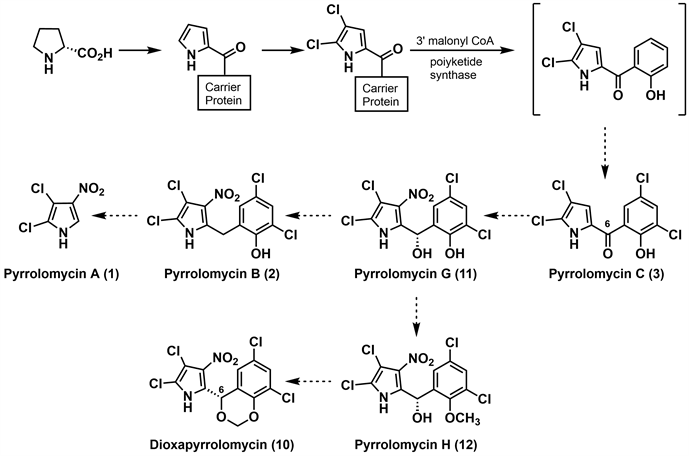
Figure 3. Parry’s hypothetical biosynthetic pathway for the pyrrolomycins [13]
图3. Parry的吡咯霉素生源合成假说 [13]
4.1.1. 吡咯霉素A和B
1981年,Ezaki等 [25] 从一株放线菌菌株的培养液(Actinosporangium vitaminophilumSF-2080)中分离得到两种广谱抗菌物质,即pyrrolomycin A(1)和pyrrolomycin B (2)。并对这两种化合物开展抗菌活性测试,结果表明其对革兰氏阳性菌、革兰氏阴性菌及一些真菌均具有良好的抑制效果,对测试的菌种最低抑制浓度(MIC)分别为≤1.56 mcg/mL、≤0.10 mcg/mL。当对JCl-ICR小鼠进行腹腔注射pyrrolomycin A(1)和pyrrolomycin B (2),其LD50值分别为21.1 mg/kg和100 mg/kg。为进一步确定pyrrolomycin A(1)的结构,Koyama等 [18] 开展了pyrrolomycin A(1)的合成研究,以3-硝基吡咯为原料,在15℃的乙酸中用等摩尔的磺酰氯氯化得到15,再加入等摩尔的磺酰氯反应最终得到收率为13%的pyrrolomycin A (1)和47%的异构体副产物及一氯取代物。这是第一次关于天然吡咯霉素的合成报道,但是存在选择性差、收率低及副产物明显等问题(如图4所示)。
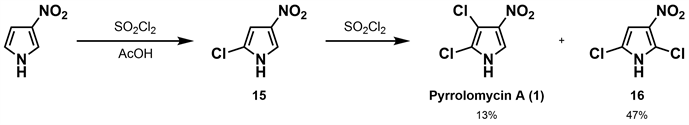
Figure 4. Synthesis of pyrrolomycin A by Koyama [25]
图4. Koyama合成吡咯霉素A [25]
4.1.2. 吡咯霉素C和D
1983年,Ezaki等 [20] [21] 从放线菌菌株的培养液(Actinosporangium vitaminophilumSF-2080)中分离得到pyrrolomycin C (3)、pyrrolomycin D (4)和pyrrolomycin E (5),并报道了其结构、物理化学特性及生物活性。根据活性研究发现,pyrrolomycin C (3)对革兰氏阳性菌具有较强的抑制活性,而pyrrolomycin D (4)具有广谱抗菌活性,对革兰氏阳性菌、革兰氏阴性菌、真菌均有抑制活性。同时pyrrolomycin C (3)和pyrrolomycin D (4)与假单胞菌产生的藤黄绿脓菌素的结构相似,但pyrrolomycin D (4)对革兰氏阳性菌和某些真菌的活性高于绿脓菌素,其对革兰氏阳性菌的最低抑制浓度(MIC)为≤0.025 µg/mL,这表明在吡咯部分引入氯原子可增强生物活性。
同年,Ezaki等 [21] 完成了pyrrolomycin C (3)和pyrrolomycin D (4)的合成工作。作者以吡咯为原料,与格式试剂反应,进一步和2-甲氧基-3,5-二氯苯甲酸甲酯反应,然后在20℃下用磺酰氯进行氯化得到中间体18和19,最后脱甲基得到收率为16%的pyrrolomycin D (4)和部分二氯取代物。二氯取代物为pyrrolomycin C (3)与其异构体混合物,后经分离纯化及鉴定pyrrolomycin C (3)的收率仅为18%(如图5所示)。该合成方法存在收率低且反应副产物较多的问题。
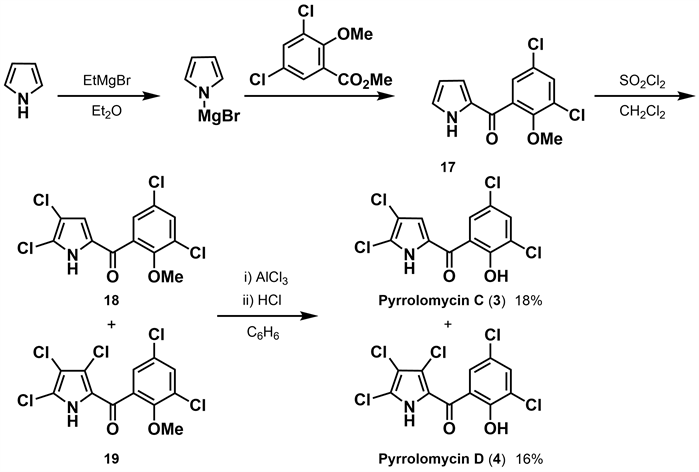
Figure 5. Synthesis of pyrrolomycin C and D by Ezaki [21]
图5. Ezaki合成吡咯霉素C、D [21]
1991年,Conder等 [26] 在Ezaki等人的研究基础上,通过降低反应温度以较高收率得到pyrrolomycin C (3)。作者通过吡咯溴化镁和3,5-二氯-2-甲氧基苯甲酰氯反应得到中间体20,在0℃条件下用磺酰氯氯化,再用AlCl3脱甲基,得到收率为51%的pyrrolomycin C (3)。在20℃条件下用磺酰氯氯化中间体20,再用AlCl3脱甲基得到收率仅为6%的pyrrolomycin D (4)。(如图6所示)通过上述方法可以实现在低温下得到较高收率的pyrrolomycin C (3),同时降低副产物的收率。根据活性研究表明,dioxapryrrolomycin (10)和pyrrolomycin C (3)对蛇形毛圆线虫(T. colubriformis)具有微弱的活性,这说明通过合成或半合成的方法提供的吡咯霉素类化合物模板可以提供新型的广谱驱虫药。
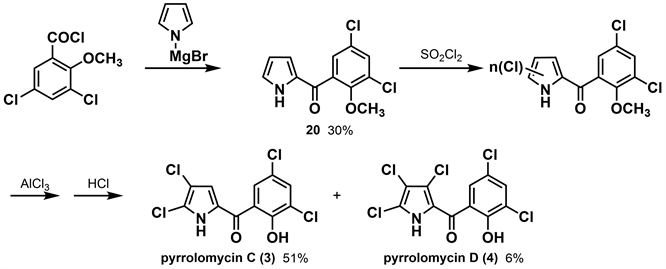
Figure 6. Synthesis of pyrrolomycin C and D by Conder [26]
图6. Conder合成吡咯霉素C、D [26]
4.1.3. 吡咯霉素F1
1983年,Ezaki等 [22] 报道了pyrrolomycin F1 (6)的合成方法,并测试了pyrrolomycin F组分的抗菌活性。作者以吡咯为原料,在无水苯和EtMgBr溶液中与5-溴-2-甲氧基苯甲酸甲酯反应,得到中间体21,再经溴化后脱甲基得到pyrrolomycin F1(6),收率为28% (如图7所示)。根据活性研究可知,pyrrolomycin F组分对革兰氏阳性菌表现出较强的活性,其中pyrrolomycin F1 (6)对金黄色葡萄球菌(Staphylococcus aureus)、粪链球菌(Streptococcus faecalis)、炭疽芽孢杆菌(Acillus anthracis)的最低抑制浓度(MIC)均为<0.5 µg/m,而对革兰氏阴性细菌抑制活性中等。根据小鼠腹腔注射试验显示,pyrrolomycin F1 (6)的LD50值为50 mg/kg。

Figure 7. Synthesis of pyrrolomycin F1 by Ezaki [22]
图7. Ezaki合成吡咯霉素F1 [22]
4.2. 新型吡咯类化合物
Dioxapyrrolomycin (10) 因具有手性和1,3-二氧六环结构而具有杀虫杀螨活性 [27]。美国氰胺公司以其为先导化合物,开发了高效杀虫杀螨剂溴虫腈(如图8)并广泛应用于防治蔬菜害虫。我国南开大学汪清民等受此启发,基于dioxapyrrolomycin和溴虫腈的结构特征,设计合成了八类新型吡咯化合物,其中三类为芳基吡咯类化合物,并开展了活性探究试验。
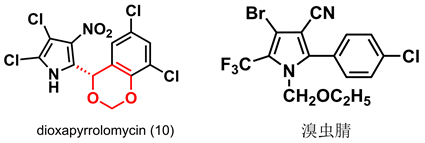
Figure 8. Schematic diagram of dioxapyrrolomycin (10) and bropononil structures [28]
图8. 二噁吡咯霉素(10)和溴虫腈结构示意图 [28]
2009年,汪清民等 [28] 基于dioxapyrrolomycin (10)和溴虫腈的结构特点,采用活性片断拼接原理,在溴虫腈结构基础上用苄基替换溴虫腈的苯基,设计并合成了新型取代苄基吡咯类化合物。作者以二苯甲酮为原料,与氨气发生亲核加成反应得到二苯甲亚胺,再与甘氨酸乙酯盐酸盐反应得到23,后与对氯苄氯反应,经过还原氧化、内酯环化等反应得到26。26与2-氯丙烯腈反应得到关键产物27,后通过溴化得到产物28,收率为84.3%。产物28与氯甲基乙醚反应得到产物29,收率为64.4% (如图9所示)。结果表明取代苄基吡咯类化合物仍然具有杀虫杀螨活性,但总体来看活性与溴虫腈相差了一个数量级。其杀虫活性与吡咯环上的溴有很大联系,没有溴原子则活性完全消失;苯环上的取代基也至关重要,对位为氯最优。具有广谱的离体杀菌活性,同时还具有一定的植物生长调节活性。
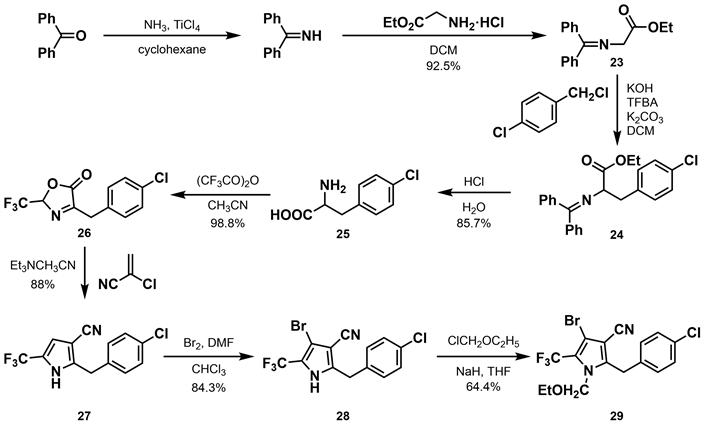
Figure 9. Synthesis of substituted benzylpyrrole derivatives [28]
图9. 取代苄基吡咯类衍生物的合成 [28]
同年,汪清民等 [28] 在前期的研究基础上开展了对苯甲酞基吡咯类衍生物31、34的合成研究。作者报道了两种合成方法,第一种(如图10所示)是以二氯甲烷为溶剂,加入二氯亚砜与30反应得到收率为91.3%的31。第二种方法(如图11所示)以三氯甲烷为溶剂,加入液溴与三氯甲烷的混合溶液与32反应得到收率为84.6%的33。再以THF为溶剂,加入氢化钠与氯甲基乙醚与33反应可得到收率为44.4%的34。
在上述两个合成中,吡咯环上的取代基除2-位外其余与溴虫腈完全相同,具有杀虫和杀螨活性 [29]。经生物活性测定证实,苯环取代基不同表现出不同程度的杀虫活性,对氯取代活性最佳。
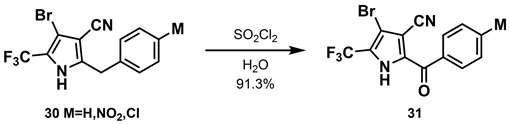
Figure 10. Synthesis of phthalide pyrrole derivatives (1) [28]
图10. 苯甲酞基吡咯类衍生物的合成(1) [28]

Figure 11.Synthesis of phthalide pyrrole derivatives (2) [28]
图11. 苯甲酞基吡咯类衍生物合成(2) [28]
4.3. 吡咯霉素衍生物
金黄色葡萄球菌(staphylococcus aureus)是造成世界各地医院和社区相关细菌感染的主要原因,且难以治疗。为了增加临床使用的抗生素种类,提供新的抗金黄色葡萄球菌和生物膜感染的药物,2016年李荣时等 [30] 采用片段和生物等排的方法首次设计和合成了一系列新型氟化吡咯霉素。李荣时等以2,3,5-三氟苯甲酸为原料,在氢氧化钠作用下邻位氟被羟基取代得到收率为95%的3,5-二氟-2-羟基苯甲酸(35)。将35甲基化、酯化得到的36与吡咯发生酰化反应,得到收率为58%的37,反应混合物中存在的MeMgI使其去甲基化,得到收率为11%的副产物38。用NBS对37进行溴代,生成收率为70%的39。39上的吡咯-NH甲基化得到收率为84%的40。用三溴化硼对39和用氯化铝对40脱甲基分别得到收率为85%的41和80%的42。(如图12所示)
对生成的化合物进行抗菌活性测试,结果表明羟基甲基化的化合物40的抗菌活性比未甲基化的41低18倍,化合物40和42的抗菌活性都比化合物41低24倍。这些结果表明,为获得有效的抗金黄色葡萄球菌活性,吡咯环上的N原子和苯环上的羟基都不能被取代。
同年李荣时等 [30] 开展了新吡咯霉素II的合成研究(如图13所示)。将二氯水杨酸酯化得到收率为95%的酯43。吡咯与酯43发生酰化反应分别得到收率为25%的44,后被MeMgI去甲基化产生22%的副产物45。用三当量或一当量的NBS溴化可以得到46和47,收率分别为29%和40%。44通过氟化反应产生了一种未经纯化的用作粗中间体的复杂混合物。经二氟化氙(XeF2)氟化、NBS溴化、AlCl3脱甲基、高效液相色谱纯化,得到了两种新型吡咯霉素48和49,未优化的收率为7%和1%。
根据活性研究试验,活性最高的氟化吡咯霉素48对金黄色葡萄球菌(Staphylococcus aureus)表现出强大的抗菌活性,clogP值为4.1,MIC值为0.073 µg/mL,MBC值为4.0 µg/mL。值得注意的是,该化合物在8.0 µg/mL的浓度下也能杀死100%的葡萄球菌生物膜,生物利用度为35%,静脉注射和口服给药的消除半衰期分别为6.04 h和6.75 h。这些有前途的新型氟化吡咯霉素证明了对金黄色葡萄球菌的有效抗菌活性,具有良好的类药物特性和药代动力学特征。更重要的是,这些化合物可以杀死葡萄球菌生物膜相关细胞,缺乏哺乳动物细胞的细胞毒性且不会产生细菌耐药性。其为开发用于治疗金黄色葡萄球菌感染与生物膜相关感染的新型抗生素提供了有前景的支架。
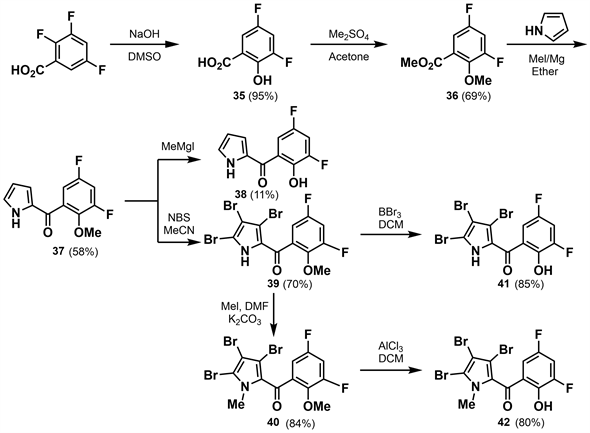
Figure 12. Synthesis of a new type of pyrrolidine fluoride I [30]
图12. 新型氟化吡咯霉素I的合成 [30]
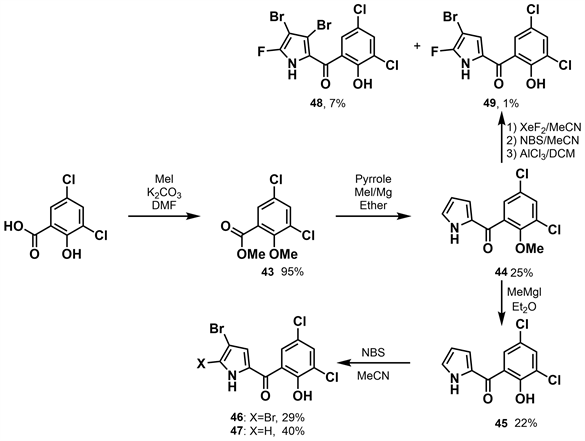
Figure 13. Synthesis of new pyrrolomycin II [30]
图13. 新吡咯霉素II的合成 [30]
5. 总结与展望
细菌耐药性作为全球卫生、食品安全和发展的重大威胁之一,耐药病原体的出现已经在全球范围内构成严重威胁,因此,迫切需要开发新的抗耐药菌的有效抗生素。吡咯霉素及其衍生物作为开发针对病原体的新型有效的潜在化合物,具有广泛的生物活性,如抗菌、杀虫、杀螨、低细胞毒活性。开展该类化合物合成及生物活性研究工作具有重要意义。
纵观吡咯霉素类化合物的合成进展,大部分集中在二十世纪八九十年代,存在合成产率低、选择性差等问题,限制了对其功能性的进一步研究。因此,发展更加高效的吡咯霉素类化合物的合成路线以及更加深入的构效关系研究,有助于设计出生物活性更高、更易制备的类似物,从而开发出抗菌效果好、安全性高且耐药性低的新型抗菌剂。
基金项目
本项目为贵州大学生“SRT计划”项目(贵大SRT字[2019]435号)和贵州省植保植检站项目(K19-0201-007号)资助。
NOTES
*通讯作者。