1. 引言
作为燃料包壳材料,Zr合金要在严苛工况中服役 [1] [2],一回路中的Zr-H2O反应将导致H的溶解和渗透 [3] [4] [5],而Zr合金中过量的H会形成脆性氢化物 [6] [7] 并影响包壳管的机械性能。计算模拟方法能在微观尺度对Zr中H的原子间相互作用和微观结构演变进行模拟,因此本文研究了运用密度泛函理论、第一性原理、分子动力学及相场法模拟Zr-H间相互作用的方法,并应用分子动力学(MD)方法自主建模并模拟了Zr-H2O体系反应过程,模拟界面处的Zr-O化学键形成过程及H扩散过程,为探究Zr-H2O界面处的Zr-H相互作用机理提供新的研究思路。
2. Zr-H相互作用计算模拟
2.1. H的吸收和扩散
已有的计算模拟结果显示,H原子优先占据四面体(TE)位 [8],且H与空位之间存在很强的相互吸引作用 [9]。hcp结构的Zr间隙位置如图1所示。
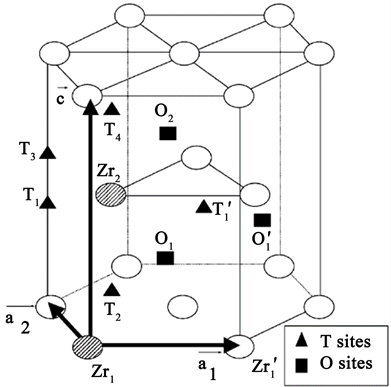
Figure 1. Labelling of interstitial sites in hcp Zr [8]
图1. hcp结构的Zr中的间隙位置 [8]
C. Domain等人 [8] 使用密度泛函理论研究了Zr-H系统的性质,通过模拟得知H原子在低温下优先占据四面体(T)位置,且H在扩散时优先沿八面体(O)位置进行,并且在基面中交替进入T位置和O位置。P.A.T. Olsson等人 [9] 通过密度泛函理论研究了氢的吸收对Zr力学性能的影响,发现氢填充的空位有助于降低表面能以及增加不稳定的堆垛层错能,使锆合金的延展性降低,且由于存在氢填充空位,断裂功和峰值应力降低。
Christopher I等人 [10] 运用LAMMPS代码进行分子动力学计算,Zr-H作用势由Christensen等人 [11] 基于EAM电位功能形式在这项工作中开发的原子间电位表示的,势函数具有以下形式:
(1)
其中,D,
和
是参数,
的值设定为5.0。通过计算得出了H在Zr中的扩散率,并与实验的测定值高度吻合,并且发现H与Zr中的空位相互作用会促进空位聚集,而H也会大大降低Zr中空位的迁移率。
2.2. ZrxHy的形成
Xueyan Zhu等人 [12] 运用第一性原理系统研究了ZrHx (x = 0.5, 1, 1.5, 2)的结构以及它们的热力学性质,发现了
结构的ZrH0.5的新稳定结构,讨论了氢化锆在不同温度下的稳定性,并提出了
的相变反应路径。计算过程中,Zr1-xHx的形成焓算式为:
(2)
其中,
、
和
分别为各物质对应的总能量。
Yongfeng Zhang等人 [13] 使用LAMMPS软件模拟了
氢化物的形成机理。由于描述Zr-H系统原子间相互作用的势函数中COMB势是最合适的 [14],因此Yongfeng Zhang等人使用COMB势 [15] 描述原子间相互作用。使用分子动力学计算了H/Zr比在1.0到2.0之间变化的各种氢化物的形成能(如图2),若所有四面体点位都被占据,则会生成
氢化物,通过随机除去H原子,可以分别以1.6和1.0的H/Zr比获得
和
氢化物,由于具有COMB电位,氢化物的fcc结构都被认为是最稳定结构,因此生成的氢化物可被视为具有各种H/Zr比的
氢化物。
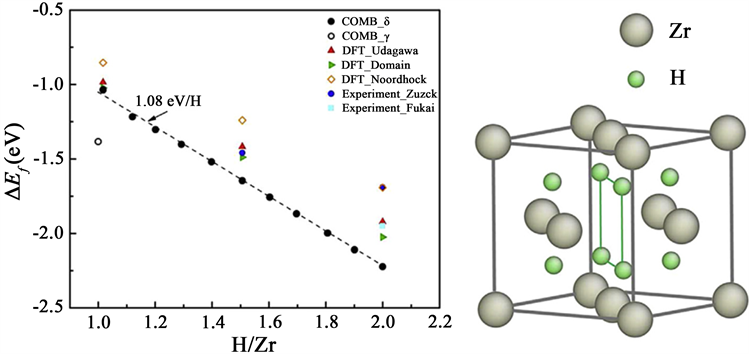
Figure 2. Hydride formation energy per ZrHX molecule as a function of the H/Zr ratio (left) and the crystal structure of hydrides used in the calculations (right) [13]
图2. ZrHx分子形成能随H/Zr比的变化趋势(左)和计算所用氢化物的结构(右) [13]
Ludovic Thuinet等人 [16] 通过密度泛函理论原理的VASP软件对Zr合金中的氢化物进行了原子级的研究,发现
-氢化物的形成可以从
-hcp或
氢化物开始,具体形成途径取决于H含量。而Yongfeng Zhang等人通过计算模拟进一步提出了
氢化物的两种可能形成的途径:1)
;2)
。两条路径都涉及hcp-fcc的相变,且最终产物都是
氢化物。
原子尺度的计算可以很好地模拟氢扩散和氢化物析出,而中尺度氢化物组织则可以通过微结构尺度的相场模拟来实现 [17]。相场模型已用于研究几种氢化物相的微观结构演变:Thuinet研究了
氢化物的微观结构的演变 [18];L. Thuinet等人 [19] 和X.Q. Ma等人 [20] 模拟了晶粒结构;外加应力情况下的
氢化物形态也由相场模拟实现 [21] [22] [23],例如X.Q. Ma等人 [21] 利用相场动力学模型研究了
-氢化物在锆中的沉淀和生长,通过数值求解结构变量的时间相关Ginzburg-Landau方程和浓度变量的Cahn-Hilliard扩散方程,从而确定空间相关场变量的时间演化,研究应力对氢化锆体系成核,生长和粗化过程的影响;Tae WookHeo等人 [24] 在近期的研究中开发了一个综合相场模型,他们所开发的模型可用于模拟包含多个晶粒
-Zr中氢化物的形成,并研究了
-Zr中的
氢化物在金属晶体中的体积膨胀现象。由下图3对比可以明显看到,相场模拟可很大程度上仿真模拟出真实情况。

Figure 3. Comparison of (a) TEM observation by Bailey [25] (b) Phase field simulation from Shi and Xiao [22]
图3. 比较(a) Bailey [25] 的TEM观察结果与(b) Shi和Xiao [22] 的模拟结果
2.3. Zr-H相互作用计算模拟方法对比
密度泛函理论仅能模拟几百个原子和短时间尺度,分子动力学模拟能模拟更大体系,并能温度和压强的共同影响;第一性原理能够通过计算实现对ZrHx的结构及其稳定性、热力学性质、氢化物相变反应路径的研究,而分子动力学能给出不同温度下的扩散系数,得到化学键断裂、结合等化学过程;相场法适用于中尺度微观模拟,可对微米量级的氢化物沉淀、生长与体膨胀现象进行计算,可与相同微观尺度的实验结果进行对比分析,模拟尺度及计算重点与分子动力学不同。应用分子动力学方法可在微观尺度下更好地模拟出一定压力、温度下Zr-H2O界面处的Zr-H相互作用,并能模拟Zr-O键的形成及H扩散过程。
3. MD模拟Zr-H2O界面处的Zr-H相互作用
Zr-H-O原子间的相互作用力决定Zr-H2O体系的微观结构与物理、化学性质,因此选用合适的原子间相互作用势是该分子动力学模拟的关键。最初认为模拟Zr-H原子间相互作用最合适的势函数是Christensen等人 [11] 基于EAM电位功能改进的EAM电势,然而EAM势并不能形成原子间化学键,因此该势函数不具备模拟Zr-H体系化学反应的能力。经过进一步调研 [13] [14] [15] 与计算模拟,确定COMB势可以完全描述Zr-H-O系统,且支持各原子间化学键的形成与断裂,适用于Zr-H2O体系的分子动力学模拟。其势函数形式为:
(3)
因此Zr-H2O体系的分子动力学模拟选用COMB3势展开。
在Zr-H2O体系模型box中,hcp结构的Zr位于box下方,H2O分子位于box上方,box尺寸为44.79 Å × 45.26 Å × 74.18 Å,H、O、Zr原子总数量设置为3884个,盒子底部是c = 5.147,a = 3.233,c/a = 1.592的hcp结构Zr基体,上部真空中随机分布100个水分子如图4所示。

Figure 4. Zr-H2O calculation model box
图4. Zr-H2O计算模型box
运用旋转矩阵R使模拟box中的H2O分子满足位置随机分布。x、y、z方向上的旋转矩阵与欧拉角旋转矩阵R如下式(4)~(7)所示:
(4)
(5)
(6)
(7)
设定box为周期性边界条件,运用NPT系综,在300~600 K的温度梯度下,运用共厄梯度法cg进行能量最小化的弛豫过程中,热力学参数时间步长2e-3ps,归一化热动力学输出值。使得到温度、电荷、压力、体积、原子位置均方位移及动势能,从微观角度研究不同工况下对锆水反应中H原子扩散的影响。
Zr-H2O体系在15 MPa、600 K的初始环境下,水分子全部分布在Zr基体(绿色)上方(图5(a)),随着模拟的进行,水分子中的H-O键(H原子为蓝色,O原子为红色)发生断裂(图5(b)),O吸附到Zr表面会形成Zr-O键,H继续向基体内扩散(图5(c))。采用共邻分析(CNA)方法,结果显示使c/a = 1.592的hcp结构数量从3136下降到2439。
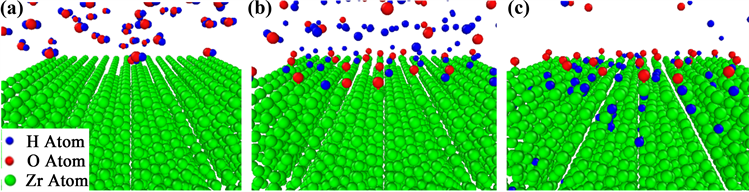
Figure 5. Zr-H2O system (a) No reaction, (b) H-O bond fracture, (c) H atom diffusion into Zr matrix
图5. Zr-H2O体系(a) 未反应,(b) H-O键断裂,(c) H原子扩散进Zr基体间隙
在运行压力15 MPa、运行温度300~600 K的工况下,随着Zr-H2O反应的持续进行,H的扩散系数D及扩散现象如图6所示,结果显示随着温度升高H扩散系数D逐渐提高。结合图6(a)~(d),可观察到温度的提高会加速H原子扩散到Zr基体,说明温度提高使锆水反应向正反应方向移动,使Zr基体体系在NPT系综下体积膨胀。

Figure 6. The H atomic diffusion coefficient of NPT at 15 MPa and different temperatures (a) 300 K, (b) 400 K, (c) 500 K, (d) 600 K
图6. NPT系综15 MPa下各温度的H原子扩散系数(a) 300 K,(b) 400 K,(c) 500 K,(d) 600 K
4. 结论
本文研究了微观尺度下Zr-H体系的计算模拟方法,得出以下结论:
1) 密度泛函理论在研究Zr中H的间隙位填充方式时具有明显优势,具备计算H在Zr中间隙固溶规律的能力。
2) 第一性原理能够通过计算实现对ZrHx的结构及其稳定性、热力学性质、氢化物相变反应路径的研究。
3) 分子动力学能够在原子尺度模拟H在Zr中的聚集与迁移行为,并能模拟Zr-H-O体系间的化学反应。
4) 相场法适用于中尺度微观模拟,可对微米量级的氢化物沉淀、生长与体积膨胀现象进行计算,可与相同微观尺度的实验结果进行对比分析。
5) 建立Zr-H2O体系反应模型,使用分子动力学方法成功模拟了界面处的锆水反应,形成Zr-O键,并模拟了H在Zr中的扩散行为,为原子尺度Zr-H2O界面处的Zr-H相互作用模拟提供了一种可行的技术手段。
基金项目
大型压水堆破损燃料检测及性状分析技术研究(KZ19011083)。
NOTES
*通讯作者。